Article Text

Abstract
Objective To report on the demographic data from the first 18 months of enrollment to an international registry on autoinflammatory diseases in the context of the Eurofever project.
Methods A web-based registry collecting baseline and clinical information on autoinflammatory diseases and related conditions is available in the member area of the PRINTO web-site. Anonymised data were collected with standardised forms.
Results 1880 (M:F=916:964) individuals from 67 centers in 31 countries have been entered in the Eurofever registry. Most of the patients (1388; 74%), reside in western Europe, 294 (16%) in the eastern and southern Mediterranean region (Turkey, Israel, North Africa), 106 (6%) in eastern Europe, 54 in Asia, 27 in South America and 11 in Australia. In total 1049 patients with a clinical diagnosis of a monogenic autoinflammatory diseases have been enrolled; genetic analysis was performed in 993 patients (95%): 703 patients have genetically confirmed disease and 197 patients are heterozygous carriers of mutations in genes that are mutated in patients with recessively inherited autoinflammatory diseases. The median diagnosis delay was 7.3 years (range 0.3–76), with a clear reduction in patients born after the identification of the first gene associated with autoinflammatory diseases in 1997.
Conclusions A shared online registry for patients with autoinflammatory diseases is available and enrollment is ongoing. Currently, there are data available for analysis on clinical presentation, disease course, and response to treatment, and to perform large scale comparative studies between different conditions.
Statistics from Altmetric.com
Introduction
The autoinflammatory diseases are a group of inflammatory conditions associated with abnormal regulation of innate immunity.1 The first molecular genetic characterisation of an autoinflammatory disease was in 1997, when two consortia independently identified the association between mutations of the MEFV (MEditerranean FeVer) gene and Familial Mediterranean fever (FMF).2 3 Over the last decade, a number of monogenic and multifactorial diseases have been identified or reclassified as autoinflammatory in aetiology1 4. With the sole exception of FMF, which is highly prevalent in some ethnicities (Turks, Arabs, non-Ashkenazi Jews and Armenians)5, the incidence of autoinflammatory diseases is low, ranging from estimated population frequencies of 1 to 3 per million for the cryopyrin associated periodic syndromes (CAPS)6 to less than 10 reported families for the most rare conditions, such as deficiency of IL-1 receptor antagonist (DIRA) or familial cold autoinflammatory syndrome 2 (FCAS2) associated with NLRP12 gene mutations.7 8
Our knowledge of autoinflammatory diseases, although expanding rapidly, is still very limited. Genotype-phenotype correlations and the potential influences of environmental and epigenetic factors are still largely unknown and, with the exception of CAPS and DIRA, their molecular pathogenesis remains elusive. Clinically, the rarity and phenotypic variability of most of these diseases means that delayed diagnosis is the norm. Even once a diagnosis has been reached we still struggle to provide patients and parents with definitive answers as to the best treatments available and their long term prognosis.
A major limitation to understanding these rare conditions has been the fragmentation of clinical experience with very few centers in any country caring for more than a handful of cases. In order to counteract this an international study, named Eurofever has been supported by the Executive Agency for Health and Consumers of European Union (EAHC, Project No2007332)9. The general aims of the project are: (i) to promote awareness and enhance early recognition of these diseases among the medical community; (ii) to provide clear and comprehensive information to families and patients affected by these conditions, (iii) to improve knowledge of the clinical presentation, response to treatment and long term complications of these disorders. The major practical part of the project was to set up an international web-based registry of autoinflammatory diseases. In this article we report on the demographic data of the patients enrolled during the first 18 months of the Eurofever registry.
Methods
A common registry for autoinflammatory diseases
The monogenic autoinflammatory diseases included were: FMF, CAPS, tumor necrosis factor -receptor associated periodic syndrome (TRAPS), Mevalonate kinase deficiency (MKD), Blau syndrome, Pyogenic Sterile Arthritis, Pyoderma Gangrenosum and Acne (PAPA) syndrome, DIRA and FCAS2. Other multifactorial autoinflammatory syndromes, such as chronic recurrent multifocal osteomyelitis (CRMO), Behcet's disease, periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis (PFAPA) and patients with undefined periodic fevers were included.
Experts in autoinflammatory diseases and syndromes were initially asked to identify the variables they considered of interest for each disease. A draft data collection form was then sent to all the experts for their review. Five main categories were considered: (i) baseline information, (ii) clinical manifestations, (iii) laboratory examinations, (iv) imaging and other diagnostic procedures and (v) response to treatments. Further revisions of the forms were subsequently evaluated by the experts with a final approval of the definitive version by nominal group technique10 during a Consensus Meeting in March 2009.
The baseline data included: age, gender, country of birth and of residence, ethnicity, age at disease onset and at diagnosis and date of first and last visits to the treating center. Genetic testing was not mandatory for inclusion. Where performed, details on the genetic analysis were sought: gene screened, extent of the analysis (specific mutations, selected exons, complete gene sequencing), mutations detected (a list was continuously updated from the Infever database, http://fmf.igh.cnrs.fr/ISSAID/infevers/)11, and laboratory where the analyses were performed. Finally, information on consanguinity and any relevant family history was collected. Patient data was anonymised and cases identified by alphanumeric codes.
Dissemination and data collection
The Eurofever project survey involved all centers linked to the Pediatric Rheumatology INternational Trial Organisation (PRINTO), a non-profit organisation devoted collaborative studies, that now encompass more than 400 centers in 60 countries worldwide12 and asked about their experience in autoinflammatory diseases. The survey results are available on the Eurofever website (www.printo.it/eurofever).
In addition, centers involved in the diagnosis and management of autoinflammatory diseases in adults were contacted and information on the study and how to participate was disseminated to members of the International Society of Systemic Autoinflammatory Diseases, the European League Against Rheumatism, the European Society of Immune-Deficiencies (ESID), through personal contacts, lectures and scientific communications and links to the Eurofever website.
The database has been developed with a sql server with through access via the member area of the PRINTO website (www.printo.it) with username and password on an https platform and URL parameters crypted. Only centers authorised (eg, centers with ethics committee approval) by the PRINTO coordinating were allowed to access the online database. Inclusion criteria for each disease were approved during the consensus conference and are reported in supplemental material. Ethical committee approval for entering patients in the registry was obtained in the participating centers as required by local legal requirements.12 Informed consent/assent as appropriate by the national laws in each country was signed by parents or legal representatives or child of adequate age. Enrollment started on November 2009.
Statistical tests were performed using subroutines from the statistical analysis package by MS Excel version 11.0.
Results
General demographic data
By data lock in July 2011, 1880 patients (M:F=916:964) from 67 centers in 31 countries have been entered in the registry, 30 centers (45%) required ethical committee approval. Enrolling countries, in alphabetic order, were: Albania, Argentina, Australia, Belgium, Croatia, Czech Republic, Denmark, France, Georgia, Germany, Greece, Hungary, India, Israel, Italy, Latvia, Lithuania, Netherlands, Oman, Poland, Romania, Russia, Saudi Arabia, Serbia, Slovakia, Slovenia, Spain, Sweden, Switzerland, Turkey and the UK.
The same Centres enrolled also patients living in other countries (Algeria, Cyprus, Egypt, Guadalupe, Iceland, Ireland, Kuwait, Lebanon, Martinique, Morocco, Norway, Uruguay, Tunisia). The distribution of the patients by country of residence is shown in figure 1. Most of the enrolled patients (1388; 74%) were resident in western Europe, 294 patients (16%) in the eastern and southern Mediterranean (Turkey, Israel, north Africa), 106 (6%) in eastern Europe (Croatia, Czech Republic, Hungary, Latvia, Lithuania, Poland, Romania, Russia, Serbia, Slovakia, Slovenia), 54 in Asia, 27 in South America and 11 in Australia. The number of enrolled patients for each disease and their distribution according to the region of residence are shown in table 1. At the time of enrollment, 1424 patients (76%) were children and 456 (24%) were adults.
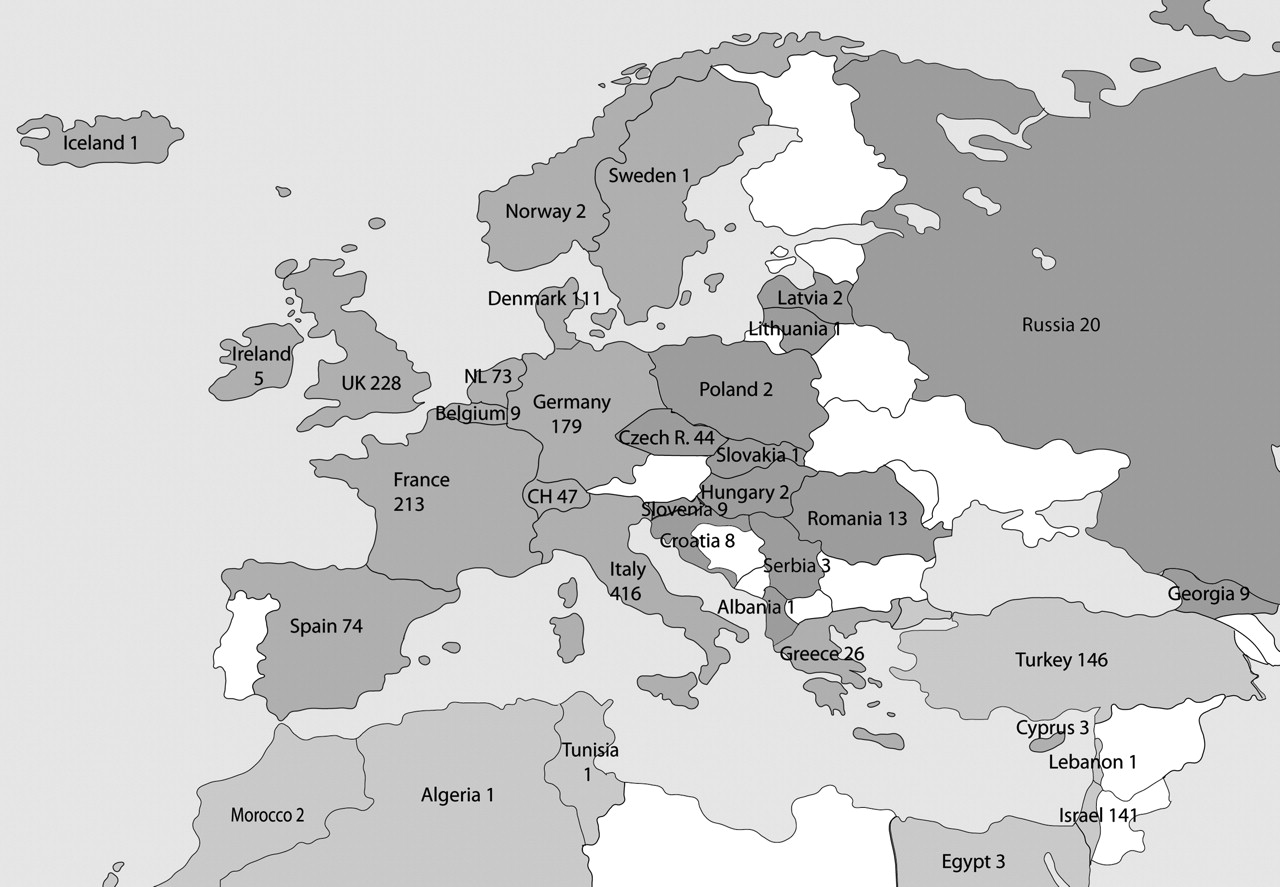
Distribution of patients according to the country of residency in Europe and the Mediterranean region. Other patients living in Saudi Arabia (33), Argentina (23), Australia (11), India (5), Oman (4), Kuwait (3), Guadalupe (2), Uruguay (1) and Martinique (1) are also reported.
Demographic characteristics of patients enrolled in the registry in the first 18 months
Monogenic autoinflammatory diseases
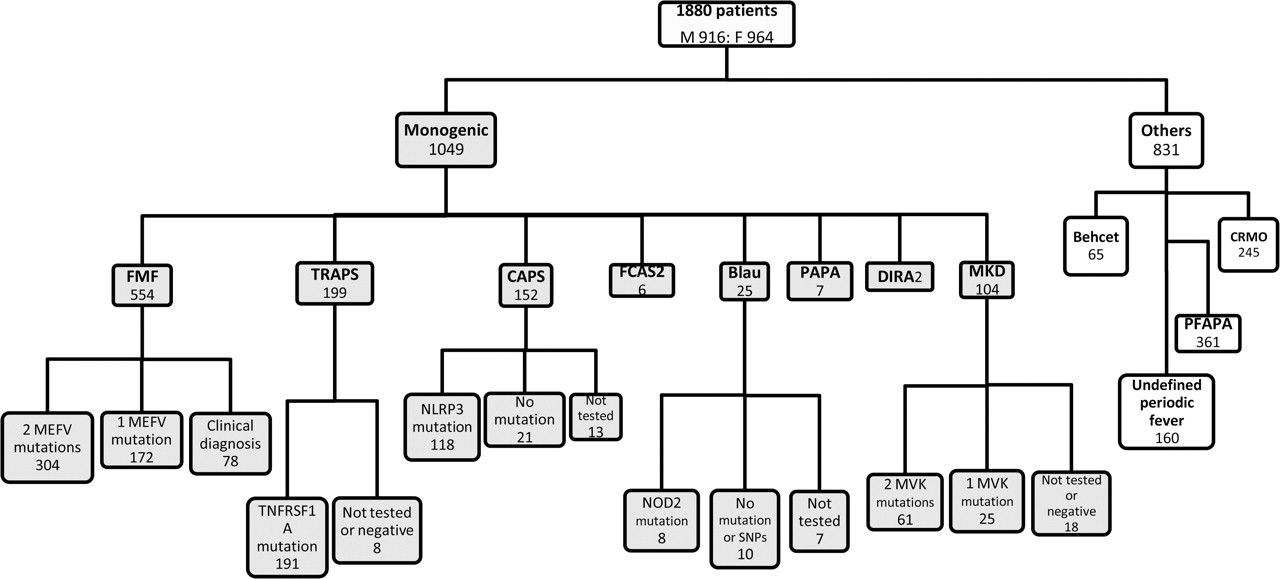
In total, 1049 patients were enrolled with a diagnosis of a monogenic autoinflammatory disease; 703 had genetic results completely consistent with the diagnosis; 197 had a heterozygous mutation in a classically autosomal recessive disease; 93 had no mutations detected and 56 patients had not been genetically tested. Most of these latter patients were not resident in western Europe and testing was not performed for technical or financial reasons. The distribution of these patients is shown in figure 2. Conceivably, the rather high rate of genetically confirmed patients was due to the selective recruitment of these patients by the centers.

Distribution of patients with autoinflammatory diseases according to disease diagnosis and genetic confirmation. FMF, familial Mediterranean fever; MKD, mevalonate-kinase deficiency; TRAPS, tumour necrosis factor receptor associated periodic syndrome; CAPS, cryopyrin associated periodic syndromes; PAPA, pyogenic sterile arthritis, pyoderma gangrenosum and acne syndrome; DIRA, deficiency of the interleukin-1-receptor antagonist; FCAS2, familial cold autoinflammatory syndrome 2; PFAPA, periodic fever, aphthous stomatitis, pharyngitis and cervical adenitis syndrome; CRMO, chronic recurrent multifocal osteomyelitis. MEFV, MEditerrean FeVer; TNFRSF1A, Tumour necrosis factor receptor superfamily member 1A; NOD2, nucleotide-binding oligomerisation domain-containing protein 2; NLRP3, NOD-like receptor 3; MVK, mevalonate-kinase; SNPs, single nucleotide polymorphisms.
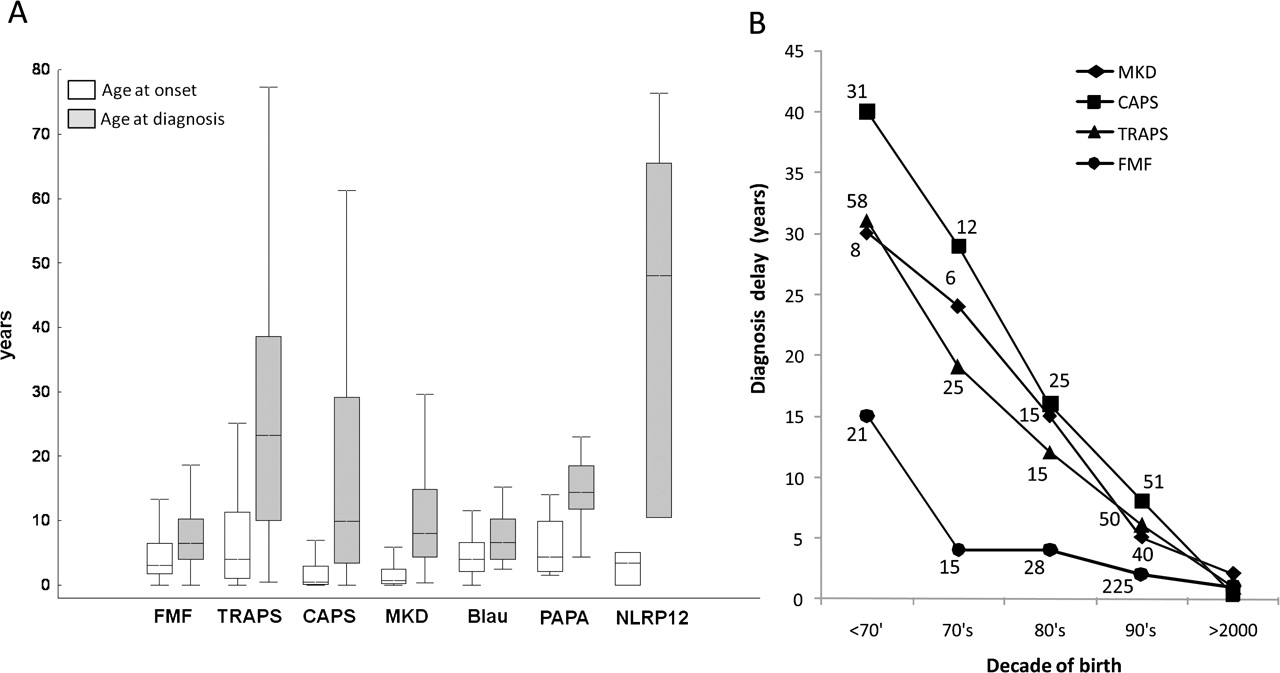
There was a marked delay between disease onset and diagnosis for all diseases – with a median of 7.3 years (figure 3A). Encouragingly there was a trend towards shorter diagnostic delay in patients born more recently (figure 3B). This was more obvious for diseases described over the last 3 or 4 decades, such as MKD, TRAPS and the more severe variant of CAPS known as CINCA/NOMID, especially after the respective molecular defects were identified.13,–,15 Nonetheless even in patients born in the 21st century the median diagnostic delay remained relatively long: 1.4 years (range 0.2–9) for FMF, 1.2 years (0.1–7.4) for TRAPS, 1.4 years (0.2–8.9) for CAPS and 2.5 years (0.1–8.3) for MKD.

{kind=link}
{kind=link}
{kind=link}
Diagnostic delay in patients with monogenic autoinflammatory diseases. The difference between the age at disease onset (white bars) and diagnosis (grey bars) is shown in panel A. Boxes contain values falling between the 25th and 75th percentiles, lines represent median values, whiskers lines that extend from the boxes represent the highest and lowest non-outlier values for each subgroups. Panel B: variation of diagnostic delay according to the decade of birth in four main inherited autoinflammatory diseases (FMF, familial Mediterranean fever; MKD, mevalonate-kinase deficiency; TRAPS, tumour necrosis factor receptor associated periodic syndrome CAPS, cryopyrin associated periodic syndrome). The number of patients are reported for each disease. For the first decade of the XXI century data on 265 FMF, 43 CAPS, 41 TRAPS and 35 MKD patients are available.
At the time of the enrollment the majority of patients (85%) were still followed by the referral centers; 638/723 (89%) children (age < 18 years) and 254/326 (78%) adults.
The distribution of the 554 FMF patients (456 children and 98 adults) according to their ethnicity and region of residence is given in table 2. Mutations found in FMF patients are reported in supplementary table I. Consanguinity was present in 78 families, and 187 patients (34%) declared a positive family history.
Baseline data for familial Mediterranean fever patients
Among the 199 TRAPS patients (85 children, 114 adults) there were 45 separate mutations, 3 of which were previously unreported (supplementary table I). Eighty-four (42%) patients reported a family history of disease; most of these carried a cysteine or the p.T50M mutation. The low-penetrance p.R92Q and p.P46L variants were reported in 75 and 7 patients, respectively (supplementary table I). The careful analysis of the clinical manifestations of this latter group of patients will help in the characterisation of the actual impact of this variant on the phenotype in comparison with mutations of the TNFRSF1A gene.16
One hundred fifty-two patients (91 children and 61 adults) with CAPS were enrolled in the registry. Among 30 NLRP3 mutations detected, 2 were previously unreported (supplementary table I). A positive family history was present in 73 (48%) patients.
There were 104 MKD patients (65 children, 39 adults) with 29 MVK variants, 4 of which were previously unreported (supplementary table I). Eight families were consanguineous and there was a positive family history in 27 (26%) patients.
Among the 25 Blau syndrome patients 8 had genetic confirmation. Following mutations were reported: p.R334Q,2 p.R334W,2 p.R587C.2 A new mutation, p.E600K, was found in 2 patients. Three polymorphisms were also found: p.P268S/SNP5 (five patients), p.R587R/SNP7 (one patient), p.R702W/SNP8 (one patient).
Six of seven patients with PAPA syndrome had confirmed mutations there were 3 previously described PSPTTP1 variants (p.A230T, p.E250K and p.E250Q) and 3 novel variants (p.V344I, p.E256G, p.G904A) non already reported in Infevers.
There were six patients with the FCAS2 associated to NLRP12 mutations; a pair of twins from Guadalupe displayed the p.R284X mutation and four Italian patients displayed the p.D294E mutation.
Finally, two patients with DIRA were enrolled from the Netherlands and were homozygous for the E77X mutation of the IL1RN gene.
Multifactorial conditions without clear genetic aetiology and undefined periodic fevers
Two hundred forty-five patients (204 children and 41 adults) with CRMO were enrolled in the registry. The oldest patient was a 61-years-old man who was diagnosed in 2010. Six families were consanguineous and there was a positive family history in five patients.
Sixty-five patients with Behçet disease (47 children and 18 adults) were in the registry. Three families were consanguineous and there was a positive family history in 14 patients.
Three hundred sixty-one PFAPA patients were entered in the registry. Although nine of them were adults, the majority of patients (88%) were less than 5-years-old at disease onset, eight patients presented after the age of 10. Five families were consanguineous and 36 other patients (10%) presented with a family history of PFAPA. One hundred sixty patients (99 children and 61 adults) were enrolled in the category of undefined periodic fever. In the majority (115, 72%), the disease onset was in childhood. Twenty-four patients (15%) presented after the age of 30. Three families are consanguineous and a positive family history was reported in 14 sporadic cases. One hundred sixteen patients (72%) were screened for at least one autoinflammatory diseases-associated gene. No mutations were found in 83%. Notably, despite the presence of low-penetrance variants, 20 patients were included this subgroup as the entering centers felt that their clinical manifestations were not consistent with disease normally associated with that gene. The various mutations found were the p.R92Q variant of TNFRSF1A (six patients); MEFV mutations in nine patients (p.R202Q,4 p.E148Q,2 p.A744S,1 p.S309F1 and p.V726A),1 the p.T356M mutation of MVK (two patients), and the p.V198M,1 p. Q703K1 and p.T219T1 variants of NLRP3.
Discussion
To be successful, a rare disease registry needs to be genuinely international and requires active ongoing support by centers involved in diagnosing and managing the patients. PRINTO, which has a track record of establishing a successful network of centers in Pediatric Rheumatology, provided a starting point for this brand new initiative in the autoinflammatory diseases. Although this EU- funded project was particularly directed at Europe the pre-existing PRINTO network facilitated expansion to other geographic areas. The broad coverage of pediatric centers was crucial because, as shown by the present study, the majority of patients with autoinflammatory diseases have an early disease onset.17 Autoinflammatory diseases are also diagnosed by adult specialists (rheumatologists, clinical immunologists and geneticists, experts in amyloidosis) and other pediatric subspecialists (immunologists, neonatologists, experts in infectious diseases). A major effort of the project was to expand the network as widely as possible by actively inviting pediatric and adult centers known to have an interest in the diagnosis and management of these patients. This was made simpler by the involvement of initiatives such as Eurotraps (http://fmf.igh.cnrs.fr/ISSAID/EUROTRAPS/) and Hyper IgD registry (http://hids.net) and international scientific societies. The ability to integrate with pre-existing registries and resources such as Infevers11 shows the power of web-based initiatives in collecting, collating and distributing data in rare diseases and in promoting networks of interested centres which can provide mutual support in diagnosis and management. Plans for further collaborations with other international and national initiatives are ongoing.18 Although a number of important geographic areas (North America, many south American and Asian countries) are not yet covered by the registry, the large number of patients recruited in this first period of the project and the wide geographical distribution is very encouraging and suggests that the network can continue to grow and prove an invaluable long term resource in the study of autoinflammatory diseases.
One of the most pressing issues in the rare diseases is delayed diagnosis. Autoinflammatory attacks are extremely unpleasant and frequently preventable with current treatments and untreated disease carries a risk of increased morbidity and irreversible long term damage. The dramatic decrease in diagnostic delay in the last decade is, to some extent, simply due to the fact that many of these diseases are recently recognised clinical entities and that genetic diagnosis has only become relatively widely available in the last few years. The first gene to be identified in autoinflammatory diseases was MEFV in 19972 3 and with identification of ILR1N in 20097 the number of genetically characterised autoinflammatory diseases reached eight. Nonetheless the diagnosis of autoinflammatory diseases relies on a clinical suspicion and many cases are missed due to poor recognition of these exceptionally rare. The uneven geographic distribution of cases reported here with a marked bias towards western Europe, where there are a relatively large number of specialist centers with facilities for genetic analysis, highlights this problem and supports the results of a survey performed among PRINTO and ESID centers which suggested that poor recognition of the autoinflammatory diseases is even more marked in the eastern European countries.19
The main aim of our project was to set-up a long term registry for the very rare monogenic diseases (TRAPS, CAPS, MKD, Blau disease, NLRP12-associated disease, DIRA, PAPA syndrome). FMF has a high prevalence in some ethnicities and a previous multi-national collaboration (Meta-FMF) has collected a large case series.20 The aim of Eurofever in FMF was not to set up a formal registry of all existing FMF patients, but rather to collect information on unselected cohorts of FMF patients of different ethnicities and residing in different regions with a view to compare them with the other monogenic periodic syndromes. A cohort of patients with multifactorial diseases which enter the differential diagnosis, such as PFAPA syndrome, CRMO and Behçet disease, were also enrolled with a view to comparative studies.
Detailed data on clinical manifestations and response to treatment are already available for more than 70% of patients. For each completed patient, an internal validation process is provided by the participating experts. The aim of this validation process is to select patients with an unequivocal genetic and/or clinical picture for future studies on genotype-phenotype correlations, complication and response to treatment. Moreover, appropriate genetic testing will be offered to patients with a strong clinical suspicion for a given disease that has not been analysed so far. The registry is still actively recruiting and new patients will be included in the follow-up of the project that will continue in the long term. Moreover, newly recognised monogenic autoinflammatory diseases will be also included.21,–,23
Establishment of a large cohort and prolonged follow-up data will allow statistically robust and clinically meaningful analysis of phenotype/genotype correlations, complications and response to treatment for most of the disease involved. Standardised collection of clinical data on both rare monogenic autoinflammatory diseases and more frequent conditions will facilitate comparison of different diseases. This will overcome the problems inherent in single disease registries when generating evidence-based diagnostic and classification criteria and/or indications for the molecular analysis24 25.
Approximately 75–80% of patients with clinical features consistent with autoinflammatory diseases have no recognised mutations in any of the known genes.24 26,–,28 A large cohort of patients with undefined periodic fevers will allow characterisation of novel genes in this currently challenging disease group. For each patient of this particular subgroup a panel of experts will evaluate the possible indications for the implementation of molecular screening for already known genes. In any case, the identification of ‘genetically negative’ patients with a well characterised clinical phenotype (ie, CAPS-like patients) and the cohort of individuals and families with an undefined periodic fever syndrome are a future resource for linkage and/or next generation sequencing analyses.29 30
Acknowledgments
The Authors would like to thank Eugenia Mosci for her outstanding and enthusiastic secretarial assistance and the webmaster Michele Pesce for his excellent work and continuous kind assistance.
References
Footnotes
-
Funding This project is supported by Executive Agency for Health and Consumers of European Union (EAHC, Project No2007332, Eurofever) and through Coordination Theme 1 (Health) of the European Community's FP7, Grant agreement number HEALTH-F2-2008-200923 (Eurotraps). Unrestricted educational grants were also kindly provided by PRINTO and Novartis.
-
Correction notice This article has been corrected since it was published Online First. The following statement was added at the end of the author list: for Paediatric Rheumatology International Trials Organisation (PRINTO), Eurotraps and Eurofever Projects.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.