Article Text

Abstract
Objective: To assess the efficacy and safety of 100 mg daily anakinra (Kineret), a recombinant form of the naturally occurring interleukin 1 receptor antagonist, plus methotrexate (MTX) in reducing the signs and symptoms of rheumatoid arthritis (RA).
Methods: Patients with active RA (n = 506) despite current treatment with MTX were enrolled in this multicentre, double blind, randomised, placebo controlled study. Patients received subcutaneous injections of anakinra 100 mg/day or placebo. They were assessed monthly for 6 months for improvement in signs and symptoms of RA and for adverse events. The primary efficacy measure was the percentage of patients attaining ACR20 response at week 24.
Results: Significantly greater proportions of patients treated with anakinra compared with placebo achieved ACR20 (38% v 22%; p<0.001), ACR50 (17% v 8%; p<0.01), and ACR70 (6% v 2%; p<0.05) responses. The response to anakinra was rapid; the proportion of patients with an ACR20 response at the first study assessment (4 weeks) was twice as high with anakinra as with placebo (p<0.005). Clinically meaningful and statistically significant responses were also seen in individual components of the ACR response (for example, Health Assessment Questionnaire, pain, C reactive protein levels, and erythrocyte sedimentation rate). Anakinra was well tolerated, with a safety profile, similar to that of placebo with one exception: mild to moderate injection site reactions were more common with anakinra than with placebo (65% v 24%).
Conclusions: This study confirms previous observations from a dose-ranging study showing that anakinra, in combination with MTX, is an effective and safe treatment for patients with RA who have inadequate responses to MTX alone.
- ACR, American College of Rheumatology
- CRP, C reactive protein
- DMARDs, disease modifying antirheumatic drugs
- ESR, erythrocyte sedimentation rate
- HAQ, Health Assessment Questionnaire
- IL, interleukin
- IL1Ra, interleukin 1 receptor antagonist
- ISR, injection site reaction
- MTX, methotrexate
- RA, rheumatoid arthritis
- rheumatoid arthritis
- anakinra
- methotrexate
- interleukin 1 receptor antagonist
Statistics from Altmetric.com
- ACR, American College of Rheumatology
- CRP, C reactive protein
- DMARDs, disease modifying antirheumatic drugs
- ESR, erythrocyte sedimentation rate
- HAQ, Health Assessment Questionnaire
- IL, interleukin
- IL1Ra, interleukin 1 receptor antagonist
- ISR, injection site reaction
- MTX, methotrexate
- RA, rheumatoid arthritis
Rheumatoid arthritis (RA) is a progressive inflammatory autoimmune disease that is characterised by proliferation of the synovial tissue and joint destruction. It results in a high level of patient disability and patients with RA have a markedly lower quality of life than the general population.1,2
The cause of RA is not known, but it is clear that activation of autoreactive T cells and macrophages is a central driving process in the disease. Macrophages amplify and maintain the pathological inflammatory response by secreting proinflammatory cytokines, such as tumour necrosis factor α and interleukin 1 (IL1). In humans, levels of IL1 in the synovial fluid correlate directly with disease activity, such that clinically active joints have much higher levels of IL1 than less active and normal joints.3 IL1 also has a fundamental role in the cartilage and bone destruction associated with RA. Plasma and synovial IL1 levels strongly correlate with erosive disease.4 In addition, patients with RA expressing autoantibodies to IL1α have a lower relative risk for joint damage than patients who do not express these antibodies.5
The naturally occurring endogenous IL1 receptor antagonist (IL1Ra) binds to IL1 receptors with an affinity similar to that of IL1α and IL1β, but does not induce a signalling response.6 This antagonist normally functions to balance the proinflammatory effects of IL1.7 An imbalance between IL1Ra and IL1 appears to be a key factor in the pathophysiology of RA. Synovial cell cultures from patients with RA produce much less IL1Ra than is necessary to inhibit IL1 activity and increased ratios of IL1Ra to IL1 correlate strongly with better prognoses in patients with arthritis.8–,10
Kineret (anakinra), a recombinant form of IL1Ra, has recently been tested in clinical trials in RA and, based on these studies, been approved for RA treatment. Significant improvements were seen in disease activity, radiographic progression, and patient reported outcomes, including functional disability, as measured by the Stanford Health Assessment Questionnaire (HAQ) and pain levels.11–,13 These clinical studies show that anakinra is an effective new treatment for RA and support the hypothesis that restoring the physiological balance between IL1 and IL1Ra by administering exogenous IL1Ra results in favourable clinical outcomes.
Methotrexate (MTX) is a standard treatment for moderate and severe RA.14 However, many patients do not respond adequately to MTX and continue to have active disease.15–,17 Previously, a dose-response study found that treatment with anakinra in combination with MTX significantly improves disease activity in patients with RA in whom there is active disease after treatment with MTX alone.13 Accordingly, we initiated a study to evaluate the effects of anakinra, given at a single 100 mg/day dose, in combination with MTX in patients with persistent disease activity after treatment with MTX alone. From findings in previous dose-response studies11,13 100 mg/day appears to be the most efficacious dose with the best safety profile. This 24 week analysis focused on American College of Rheumatology (ACR) responses18 and patient reported outcomes as the efficacy measures.
PATIENTS AND METHODS
Patients
Patients were eligible for the study if they were at least 18 years old; had a diagnosis of RA according to the ACR criteria19; had disease duration of at least 24 weeks before study entry; had radiographic evidence of bone erosion in the hands, wrists, or feet; and had active RA. Active RA was defined as six or more swollen joints, nine or more tender or painful joints, and either a C reactive protein (CRP) level of at least 15 mg/l or an erythrocyte sedimentation rate (ESR) of at least 28 mm/1st h. Patients were also required to have been treated with a stable dose of MTX (10–25 mg/week) for at least 24 consecutive weeks. Patients who were taking MTX (25–50 mg) every 2 weeks were also eligible for the study. Patients who were taking non-steroidal anti-inflammatory drugs or oral corticosteroids (⩽10 mg/day of prednisone equivalent) were eligible if the dose had been stable for at least 4 weeks before randomisation.
Patients were excluded from the study if they had any clinically significant systemic disease or autoimmune disease other than RA, had a serious infection, leucopenia, or allergy to products derived from Escherichia coli, or were being considered for surgery to their hands, wrists, or feet. Patients were also excluded if they had been treated with intra-articular or systemic corticosteroid injections within 4 weeks before the study, or were being treated with disease modifying antirheumatic drugs (DMARDs) other than MTX (washout period of 60 days before randomisation), or required narcotic analgesics for pain, or had been treated previously with IL1Ra.
Written informed consent to participate in the study was obtained from each patient. The study was conducted according to the principles of the Declaration of Helsinki, FDA regulations, and International Conference on Harmonisation and Good Clinical Practice guidelines.
Study design
This study was a 24 week multicentre, double blind, randomised, placebo controlled study that was the first part of a larger 52 week study designed to evaluate the effects of anakinra on radiographically observed structural damage in RA. Each part of the study was prospectively designed with its own primary end point: ACR responses for the 24 week study and assessments of radiographic damage for the 52 week study.
Patient eligibility was determined during an initial 21 day screening period. Eligible patients were randomised to single daily subcutaneous injections of anakinra (100 mg) or placebo. All patients were treated with concurrent MTX (10–25 mg/week) and were required to take folic acid (1 mg) daily. The injections of anakinra or placebo were administered at home by the patient or a care giver, except on the assessment days, when they were administered at the clinic. One week after beginning treatment, patients were assessed for adverse events. Every 4 weeks a “blinded” evaluator assessed patients for disease activity and adverse events. Owing to an early change in study design, which increased the number of patient evaluations to include assessments at weeks 4 and 8, a small number of patients receiving the study drug (n = 74) were not assessed at both of these times.
Clinical measurements
The primary efficacy end point was the proportion of subjects who attained an ACR20 response at week 24.18 Secondary efficacy end points included the change from baseline at week 24 in individual ACR components, including patient’s assessment of disease activity, patient’s assessment of pain, HAQ score, plasma CRP level, and ESR; ACR50 and ACR70 responses; and sustainability of the ACR20 responses.
The number of swollen joints was determined by examining 66 diarthrodial joints for synovitis or periarticular swelling. The number of tender or painful joints was determined by examining 68 diarthrodial joints for tenderness and pain. Joints were assessed by independent evaluators who were blinded to the presence of injection site reactions (ISRs). Patient and physician assessments of disease activity and patient assessments of pain were evaluated by visual analogue scales (0–100). Each patient’s level of functional disability was evaluated using the HAQ disability score (0–3). Plasma CRP levels were measured using standard methods by the Covance Central Laboratory Services Inc (Indianapolis, IN, USA). ESR was measured locally at each study site and efforts were made to keep the results blinded to study site personnel.
Safety was evaluated by assessing adverse events and serious adverse events, including serious infections, malignancies, and deaths, and details were recorded by the study investigators. Serious adverse events were any events that suggested a significant hazard or side effect. These included events that were fatal or life threatening (placing the patient at immediate risk of death), necessitated admission to hospital or prolongation of an existing hospitalisation, or caused a persistent or significant disability or incapacity. Important medical events that jeopardised a patient, required intervention to prevent any of the above outcomes, or resulted in urgent investigation were also considered serious (for example, convulsions).
Statistical analysis
All study results, with the exception of the completers analysis (see below), are reported for the intention to treat population, which included all randomised patients who were given at least one dose of the study drug. Patients whose ACR response status could not be ascertained, because of either a missing evaluation or an inability to fully evaluate the response because of missing ACR components, were classified as non-responders for the relevant time point. In addition, any subject who had an increase from baseline in the dose of DMARDs or corticosteroids during the study was considered to be a non-responder on or after the first occurrence of the increase in these drugs. The sample size for the 24 week assessment of the ACR20 composite end point was prospectively chosen to be 500 patients, with 250 patients randomised to each treatment arm. Given a two tailed α level of 0.05, the analysis would have 90% power to detect a difference of 13% or more in the proportion of ACR20 responders between the placebo and anakinra groups.
The analysis for the ACR response rates at week 24 was a logistic regression model adjusted for study centre. A sensitivity analysis, the completers analysis, was conducted to examine the effect of missing signs and symptoms data on the week 24 ACR20 response rates in each treatment group. In this analysis, patients (23%) with missing ACR20 response data at week 24 because of early withdrawal from the study, a missed visit, or incomplete assessments were excluded from the analysis.
Analyses similar to those conducted for the ACR20 responses were also conducted for the sustained ACR20 responses. Subjects who had a sustained ACR20 response had ACR20 responses for at least 4 (not necessarily consecutive) of the 6 months, with at least one response being achieved at week 12 or 24. For patients who missed their 4 and 8 week assessments, a sustained ACR response was defined as a response in at least 3 of the 4 months in which they were assessed.
For each ACR component, the mean change from baseline at week 24 was analysed by using least squares mean values obtained from a repeated measures mixed model analysis of covariance20 adjusted for centre and the corresponding baseline value. The rates of adverse events and serious adverse events were tabulated by treatment group. All statistical analyses were performed at a two tailed α level of 0.05.
RESULTS
Patient demographics and pretreatment characteristics
To achieve the target sample size of 500 patients, all patients randomised on or before 18 May 2000 were selected. This resulted in a total of 506 patients evenly randomised to the anakinra (n = 253) and placebo (n = 253) groups. Three patients in the anakinra group and two in the placebo group were not given any study drug and thus are not included in the analyses. The final intention to treat population consisted of 501 patients (anakinra = 250, placebo = 251). Patient demographics and disease status at study entry were well balanced between the treatment groups (table 1⇓) and were typical of those in patients with active RA.
Baseline patient characteristics
Table 2⇓ summarises the components of the ACR response and the duration of morning stiffness at study entry. There were no clinically meaningful differences in RA status between the anakinra and placebo groups at study entry. These values are typical of those in patients with moderate to severe RA.
Baseline disease characteristics
ACR responses
The reduction in the signs and symptoms of RA as measured by the ACR20 response was clinically and statistically significantly greater in the anakinra group than that in the placebo group at 24 weeks (fig 1A⇓). An improvement of at least 20% in the ACR composite score (ACR20 response) was seen in a significantly greater proportion of patients in the anakinra group than in the placebo group, 38% v 22% (p<0.001). The odds ratio for a response in the anakinra group relative to the placebo group was 2.36 (95% confidence interval (CI) 1.55 to 3.62). A sensitivity analysis was conducted for patients who had non-missing ACR composite scores (completers analysis) at week 24 (fig 1B⇓). The results are nearly identical to those of the primary analysis.

(A) Proportions of patients in the anakinra (n = 250) and placebo (n = 251) groups with ACR20, ACR50, and ACR70 responses at the end of the 24 week study period. (B) Proportions of patients completing the study in the anakinra (n = 200) and placebo (n = 187) groups with ACR20 responses at the end of the 24 week study period. (C) Proportions of patients in the anakinra and placebo groups with an ACR20 response, by study week. (D) Proportions of patients in the placebo and anakinra groups with a sustained ACR20 response, defined as an ACR20 response in at least four of the six monthly assessments. Comparisons between study drugs shown as *p<0.05, **p<0.01, and ***p<0.001.
Similarly, significantly greater proportions of patients in the anakinra group than in the placebo group achieved ACR50 and ACR70 responses (see fig 1A⇑). Patients receiving anakinra were more than twice as likely to achieve an ACR50 response at week 24 (17%) than patients receiving placebo (8.0%; p<0.01), with an odds ratio of 2.61 (95% CI 1.46 to 4.84). An ACR70 response at week 24 was seen in 6% of patients in the anakinra group and in 2% of patients in the placebo group (p<0.05), with an odds ratio of 3.14 (95% CI 1.16 to 10.06).
Time course of ACR20 response
Anakinra had an early onset of action, as shown by the ACR20 response (fig 1C⇑). Approximately twice as many patients in the anakinra group (21%) as in the placebo group (11%) had an ACR20 response at the first study assessment, at week 4 (p<0.005). The proportion of patients in the anakinra group with an ACR20 response continued to increase up to week 20, to a value of 38% (p<0.001).
Sustainability of ACR20 response
Patients in the anakinra group were more than twice as likely as those in the placebo group to have a sustained ACR20 response (fig 1D⇑). A sustained ACR20 response was seen in 27% of the anakinra group v 12% of the placebo group (p<0.001), with an odds ratio for a sustained response of 3.43 (95% CI 2.05 to 5.90) in favour of anakinra over placebo.
Components of the ACR response
Improvement in individual components of the ACR composite response was seen in patients treated with anakinra. Statistically significant improvement was observed for all components except for swollen joint count (fig 2⇓). Physician’s global assessment of disease activity was significantly improved in patients treated with anakinra compared with those given placebo (−25.2 v –20.1; p<0.05) as was tender/painful joint count (−12 v –8.7; p<0.01) (fig 2⇓). An improvement of 6.8 in the swollen joint count was seen in patients treated with anakinra, relative to baseline, but placebo treated patients also improved (6.5) (table 3⇓). As noted in previous anakinra studies, significant improvement compared with placebo was demonstrated in patient reported outcomes. Patient rating of disease activity, pain, and level of disability, as measured by the HAQ score (fig 2⇓, table 3⇓) all showed significant improvement.
Change from baseline in ACR components at 24 weeks

(A) Changes in physician assessment of patient disease activity in the anakinra (n = 250) and placebo (n = 251) groups, by study week. (B) Changes in swollen joint count in the anakinra (n = 250) and placebo (n = 251) groups. (C) Changes in tender/painful joint count in the anakinra (n = 250) and placebo (n = 251) groups. (D) Changes in patient perceptions of overall disease activity in the anakinra (n = 250) and placebo (n = 251) groups. (E) Changes in patient ratings of pain in the anakinra (n = 250) and placebo (n = 251) groups. (F) Changes in patient assessments of functional disability, as measured by the HAQ, in the anakinra (n = 250) and placebo (n = 251) groups. The dashed line indicates the minimal clinically important difference in score. Comparisons between study drugs is shown as *p<0.05, **p<0.01, and ***p<0.001.
As early as week 4, patients receiving anakinra began to show more improvement in HAQ scores than those receiving placebo. By week 24, patients treated with anakinra showed a marked improvement relative to placebo patients on the HAQ (0.29 v 0.18; p<0.05). This change of 0.29 is well above the threshold of 0.22 determined to be clinically meaningful.21 Overall, 100 patients receiving anakinra reached clinically meaningful improvement (HAQ decreases ⩾0.22) at week 24 compared with 83 patients taking placebo.
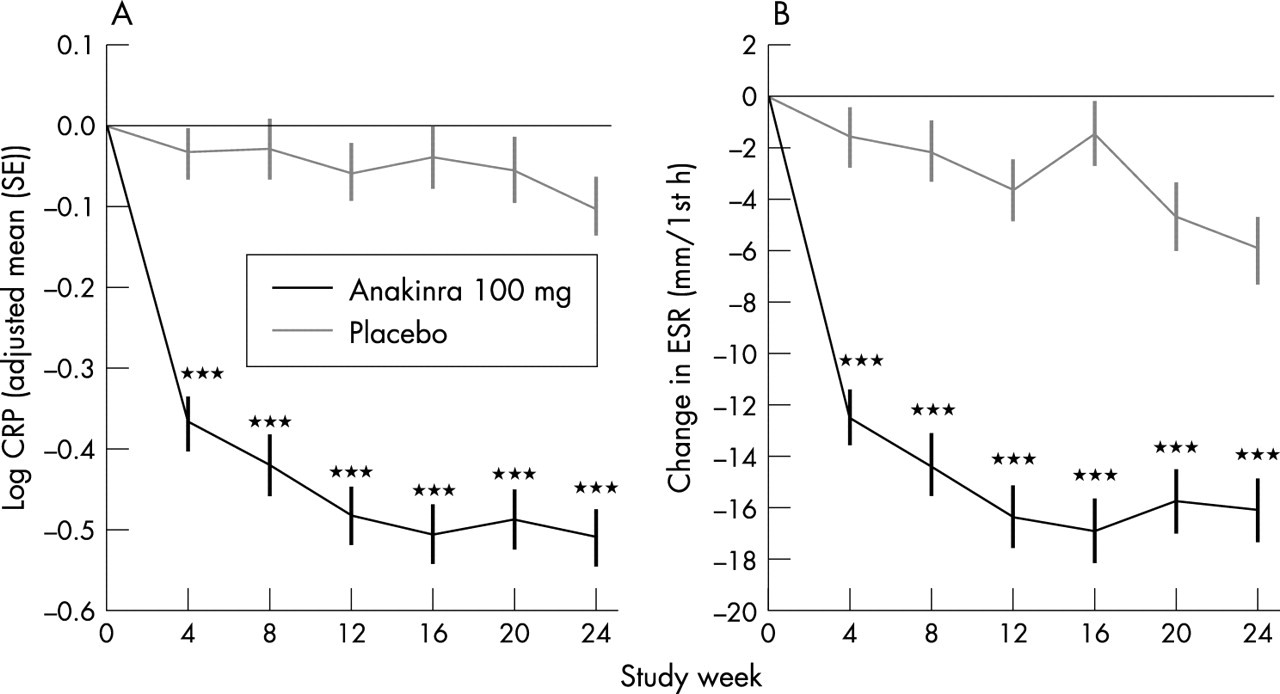
Patients in the anakinra group also showed a marked improvement, relative to those in the placebo group, in the objective end points of changes in CRP and ESR values (fig 3⇓). The change in the log transformed CRP level with anakinra was −5 and with placebo −1 mg/l (p<0.001). The improvement in the ESR with anakinra was also significantly greater than the change with placebo (−16.2 v –6.0 mm/1st h; p<0.001). Improvements in these acute phase reactant values were apparent by the week 4 assessment and were maintained throughout the study.
{kind=link}
{kind=link}
{kind=link}
(A) Log transformed changes in CRP levels in the anakinra (n = 250) and placebo (n = 251) groups at each study assessment. (B) Changes in ESR in the anakinra and placebo groups at each study assessment. Comparisons between study drugs shown as ***p<0.001.
Adverse events
Table 4⇓ summarises the safety profiles of patients treated with anakinra and placebo in this study. Overall, adverse events occurred in a similar number of patients in the anakinra (90%) and placebo (81%)groups. ISRs were more common in the anakinra group than in the placebo group (65% v 24%). Twenty one (8.4%) patients in the anakinra group and two (0.8%) patients in the placebo group withdrew from the study owing to ISRs. However, most ISRs were mild to moderate and none were considered to be serious adverse events. Most patients had their first ISR during the first month of the study and the risk of experiencing an ISR for the first time after the first month of treatment was low.
Adverse events
Similar frequencies of other adverse events were reported in patients treated with anakinra and placebo. Additionally, the overall rate of serious adverse events (anakinra 4%; placebo 3%) as well as serious adverse events that led to study withdrawal (anakinra 0.8%; placebo 1%) occurred at similar rates in both treatment groups. Serious adverse events leading to study withdrawal in the placebo group included gangrene, fracture, and prostate carcinoma and in the anakinra group interstitial lung disease that was accompanied in one patient by pulmonary fibrosis.
Infectious events occurred at a similar rate between the anakinra (33%) and placebo (26%) groups. The most common infectious events were in the respiratory tract (21% with anakinra v 16% with placebo). Infectious events in other body systems occurred in 15 or fewer patients in each treatment group. Serious infections were rare and occurred in 0.8% of both treatment groups.
DISCUSSION
This study demonstrates that the addition of anakinra (100 mg/day) to patients with persistent and active disease despite MTX treatment results in statistically significant improvement in signs and symptoms of RA as measured by the ACR response criteria. Approximately twice as many patients had ACR20, ACR50, and ACR70 responses with anakinra plus MTX as did those treated with placebo plus MTX. In addition, laboratory measures indicative of systemic inflammation were significantly decreased in the anakinra group, as evidenced by a fivefold greater decrease in the log transformed CRP levels with anakinra than with placebo and a threefold greater decrease in the ESR. These data confirm the observations reported in a previous dose-response study of anakinra plus MTX for RA treatment.13 The majority of patients in that study at those dose levels were receiving 100 mg/day or less. Based on that information, a favourable risk-benefit ratio, and the need to develop a biologic therapy that is easily administered, we elected to test 100 mg daily as a fixed dose. The results presented in this paper support the use of this once daily dose for RA treatment.
A rapid response to anakinra similar to that found in previous randomised clinical trials11,13 was seen with this 100 mg/day dose, and this response was sustained compared with the response observed in placebo patients. Improvements in the individual components of the ACR composite response were seen in patients treated with anakinra and these were statistically better than with placebo for all components except for swollen joint count. Improvements were seen in the swollen joint count for patients treated with anakinra but patients receiving placebo showed a similar improvement.
Previous studies have demonstrated a significant impact of anakinra on patient reported outcomes, including pain, disability, and rating of disease activity.11,13 Anakinra monotherapy has also been shown to improve patient functioning as indicated by fewer missed days of work22 and improved health related quality of life.23 An improvement in patient reported outcomes was seen in the current study and suggests that anakinra is effective at treating these features of RA.
The dose of anakinra (100 mg/day) tested in the current study was well tolerated, with no clinically meaningful differences between the treatment groups in serious adverse events, adverse events that led to study withdrawal, or serious infections. ISRs were more common in the anakinra group; however, none were classified as serious. Mild to moderate ISRs are known to be associated with the subcutaneous administration of anakinra, and their frequency in this study is consistent with that found in previous studies.13,15 In addition, most patients had their first ISR during the first month of the study, and the risk of experiencing an ISR for the first time after that month was low.
In this study the overall incidence of infectious episodes was reported more often in patients treated with anakinra (33%) than in placebo treated patients (26%), similar to previous trials evaluating other biologic therapies.24 The incidence of serious infectious episodes, however, was the same for both treatment groups (0.8%). This result contrasts with the higher incidence of serious infectious episodes (2.1% anakinra; 0.4% placebo) reported in a 6 month study of 1399 patients.25 As with tumour necrosis factor α inhibitors infectious episodes are seen more frequently in patients with comorbidities such as diabetes, chronic lung disease, and concomitant corticosteroid treatment.
Studies with anakinra monotherapy have shown that anakinra treatment lessens joint destruction.11,12 Specifically, over a 6 month treatment period, patients treated with anakinra showed significantly less radiographic progression as measured by decreased joint space narrowing (58% reduction) and greater reductions in the numbers of erosions (38%–46% reduction) than did placebo treated patients. Although radiographic assessments were not made in this study, the 12 month radiographic end point will be reported at the end of the study after data analysis.
In summary, this study, in a large population of patients with RA, shows that anakinra 100 mg/day in combination with MTX is safe and efficacious in patients with active disease despite MTX monotherapy. As it is most likely that anakinra, similar to other new treatments, will be primarily used in combination with DMARDs such as MTX, this study confirms the previous observations of efficacy and safety of this combination.
Acknowledgments
We are indebted to the 145 principal investigators and their study coordinators for conducting this study, and to Holly Strausbaugh, PhD, for editorial assistance.
This study was supported by Amgen, Thousand Oaks, CA, USA.