Article Text

Abstract
OBJECTIVE Recent studies have demonstrated the short term efficacy of leflunomide. This study evaluates the efficacy and safety of leflunomide and sulfasalazine in rheumatoid arthritis over a two year follow up period.
METHODS 358 patients with rheumatoid arthritis in a double blind trial were randomly allocated to receive either leflunomide 20 mg/day, placebo, or sulfasalazine 2 g/day. Those completing six months of treatment (n=230) were given the option to continue in 12 (n=168) and 24 (n=146) month double blinded extensions; the placebo group switched to sulfasalazine. This report compares efficacy and safety of leflunomide with sulfasalazine in the 6, 12, and 24 month patient cohorts.
RESULTS The efficacy seen at six months was maintained at 12 and 24 months. Twenty four month cohorts on leflunomide showed significant improvement compared with sulfasalazine in doctor (−1.46 v−1.11, p=0.03) and patient (−1.61 v−1.04, p<0.001) global assessments, ACR20% response (82%v 60%, p<0.01), and functional ability (Δmean HAQ −0.65 v −0.36, p=0.0149; ΔHAQ disability index −0.89 v −0.60, p=0.059). Improvement in other variables was comparable for the two drugs, including slowing of disease progression. Improved HAQ scores in 6, 12, and 24 month leflunomide cohorts were seen in both non-responders (24%, 29%, 35%, respectivelyv sulfasalazine 8%, 10%, 27%) and ACR20% responders (leflunomide 63%, 62%, 66% vsulfasalazine 50%, 64%, 44%). Leflunomide is well tolerated at doses of 20 mg. No unexpected adverse events or late toxicity were noted during the two year period. Diarrhoea, nausea, and alopecia were less frequent with continued treatment.
CONCLUSION These long term data confirm that leflunomide is an efficacious and safe disease modifying antirheumatic drug.
- randomised trials
- rheumatoid arthritis
- leflunomide
- sulfasalazine
Statistics from Altmetric.com
Rheumatoid arthritis (RA), which affects 1% of the population world wide, is associated with substantial premature mortality and morbidity. The enormity of the socioeconomic burden of musculoskeletal disorders, including RA, has precipitated the declaration of the years 2000–2010 as the “Bone and Joint Decade,” a global effort to contain the ever increasing costs of caring for patients with RA and aiming at optimising RA treatment modalities.1 Traditional treatment of RA has centred on the use of non-steroidal anti-inflammatory drugs (NSAIDs), and in more severe cases, judicious use of corticosteroids and disease modifying antirheumatic drugs (DMARDs). Over the past decade there has been a concerted effort aggressively to control disease activity during the early stages of the disease using a single or combination DMARD regimen. Evidence suggests that consistent use of DMARDs delays health related functional disability.2 However, most of the current DMARDs can seldom be given for long periods because of toxicity or lack of sustained efficacy.3-5 There is therefore a need for new effective agents that can provide high efficacy with minimal safety issues.
Leflunomide, an isoxazole derivative and inhibitor of de novo pyrimidine synthesis,6 ,7 represents a new class of DMARDs. The efficacy and safety of leflunomide in RA have been demonstrated in phase II and III studies of patients with RA.8-11 The primary mode of action of leflunomide is specific inhibition of dihydro-orotate dehydrogenase, a key enzyme in the de novo synthesis of pyrimidine, and subsequent inhibition of RNA and DNA synthesis.12 ,13 Activated T lymphocytes, which predominantly synthesise pyrimidines via the de novo pathway,14 may be especially susceptible to leflunomide. The immunomodulatory and anti-inflammatory effects of leflunomide have recently been reviewed by Breedveld and Dayer15 and include blockade of tumour necrosis factor mediated activation of the transcription factor NFκB16; inhibition of reactive oxygen radicals17; inhibition of polymorphonuclear leucocyte chemotaxis and the migration of polymorphonuclear leucocytes into the rheumatoid synovial cavity18; and inhibition of matrix metalloproteinases (MMPs) and subsequent increases in tissue inhibitor of metalloproteinase (TIMP)/MMP ratios in vitro19 and in patients with RA.20
In a double blind, randomised trial of leflunomide compared with placebo and sulfasalazine in patients with active RA9we have previously reported that leflunomide treated patients were significantly improved at six months , which was better than placebo and comparable with sulfasalazine. In the current report, we extend these observations and report better and, in some variables, significant results after a follow up of two years with leflunomide compared with sulfasalazine.
Methods
STUDY SUBJECTS
The study group comprised consenting patients ⩾18 years of age with active RA based on American College of Rheumatology (ACR) criteria and ACR functional class I, II, or III. Inclusion criteria at study entry included tender joint count ⩾6; swollen joint count ⩾6; doctor and patient global assessment as fair, poor, or very poor; C reactive protein (CRP) >20 mg/l or erythrocyte sedimentation rate (ESR) >28 mm/1st h. Women who were pregnant, breast feeding, or of childbearing potential not taking oral contraceptives were excluded.
Permitted concomitant drugs included stable doses of NSAIDs, including acetylsalicylic acid, oral steroids (prednisolone ⩽10 mg/day), and up to three intra-articular steroid injections, not exceeding an equivalent dose of 60 mg triamcinolone. Intra-articular steroid injections were not permitted during the first six months.
STUDY DESIGN
The study design was a multicentre, multinational (sites in Australia, Europe, New Zealand, and South Africa), randomised, double blind, placebo controlled (until six months), parallel group study. A one week screening period was followed by 24 months of treatment with leflunomide (100 mg a day on days 1–3; then 20 mg a day) or sulfasalazine (0.5 g, 1.0 g, and 1.5 g a day, respectively at weeks 1, 2, and 3; 2.0 g a day from week 4 to the end point), and matching placebo for six months. After six months, completers elected to continue treatment for up to 12 or 24 months. Patients in the leflunomide and sulfasalazine arms continued to receive the same dosage, whereas those in the placebo arm switched to sulfasalazine in a blinded manner. Patients in the placebo-sulfasalazine arm received forced-dose escalation to 2.0 g. A total of 358 patients were randomly allocated (3:3:2) to receive leflunomide (n=133), sulfasalazine (n=133), and placebo (n=92). Of the 230 patients completing six months of treatment, 168 and 146 patients, respectively, opted to participate in 12 and 24 month double blinded extensions. Efficacy and safety was assessed in 6, 12, and 24 month patient cohorts.
EVALUATION CRITERIA
Efficacy was assessed at baseline, at two week intervals for the first two months, at monthly intervals for months 2–6, every three months for months 6–12, and every two months until the end point. Efficacy outcomes included tender and swollen joint counts, doctor and patient global assessments, pain intensity assessment, duration of morning stiffness, Westergren ESR, CRP, rheumatoid factor (RF), and functional disability. Data on functional disability were assessed by the Health Assessment Questionnaire (HAQ)2 in two ways: (a) as the mean HAQ scores of all eight categories, where each category score represents the arithmetic mean of the item scores for that category,21 ,22 and (b) as the standard HAQ disability index, where the category score represents the worst item score for that category. Also assessed was the ACR responder rate (defined as ⩾20% improvement from baseline in tender and swollen joint counts and ⩾20% improvement in three of the following five criteria: doctor global assessment, patient global assessment, pain intensity assessment, physical disability, and CRP/ESR). The proportions of patients with 50% and 70% ACR response were also calculated.
Disease progression was evaluated by radiographic analysis of both hands and feet. Radiographs were obtained at baseline and at months 6, 12, and 24. Radiographs pertaining to the 0–6 month cohorts, which were blinded for treatment and sequence, were read by a single observer using the original Larsen method23 as applied to clinical trials.24 Radiographs pertaining to the 0–12 and 0–24 month cohorts were also evaluated by a single observer who was unaware of the treatment and subject but not the sequence.
SAFETY
Safety was monitored by physical examination, chest radiography, electrocardiography, and blood pressure, pulse rate, body temperature, and body weight measurements. Standard haematological and biochemical tests and urine analysis were done. The occurrence of adverse events (AEs) was recorded and included those spontaneously reported by patients, as well as those elicited by general questioning. Investigators were instructed to record AEs as primary events (that is, the diagnosis of an isolated event) and symptoms accompanying the primary event. Analyses of the AEs were done on the basis of the primary events. A serious AE was defined as fatal, life threatening, permanently disabling, necessitating hospital admission, congenital anomaly, cancer, or overdose. Laboratory variables were analysed at a central laboratory, and results are presented as median values at baseline and end point.
STATISTICAL ANALYSES
All analyses reported were performed on the intention to treat population. This study was not powered to show equivalence between the active treatment arms, and 95% confidence intervals were used to describe the magnitude of the differences. Mean changes from baseline to end point between treatment groups were analysed by pairwise comparisons between treatment groups for all continuous variables, including HAQ analysis using the technique of last observation carried forward and analysis of covariance (adjusting for centre effects, disease duration, and baseline value). Patient and doctor global assessments were compared by the extended Mantel-Haenszel test. All laboratory variables were subjected to descriptive statistics, and changes between baseline and end point were compared within treatment groups by the Wilcoxon signed rank test. Rates of ACR response were analysed by logistic regression, adjusted for centre effects and disease duration. All statistical tests were two sided, and the significance level was defined as p<0.05.
Results
BASELINE FEATURES
The three treatment groups had similar baseline demographic characteristics (table 1). Their mean ages varied from 58 to 59 years and their mean disease durations from five to eight years. Between 38% and 46% of cases had disease duration of two years or less. Most patients were of ACR functional class II or III, and 39–53% had not previously received DMARD treatment. A high proportion of patients (72–85%) used NSAIDs, and corticosteroid use was similar (32–52%).
Baseline demographics. Results are given as No (%) unless otherwise staed
SEGMENTS OF THE STUDY
Of 358 randomised patients, 133 receiving leflunomide, 92 placebo, and 133 sulfasalazine participated in the first six month study period (fig 1). Of the 230 who completed six months of treatment, 197 patients (80 leflunomide and 76 sulfasalazine treated, and 41 patients in the placebo group who switched to sulfasalazine) entered the 6–12 month extension. Of the 168 patients who completed 12 months of treatment, 146 (60 leflunomide, 26 placebo-sulfasalazine, and 60 sulfasalazine treated patients) entered the 12–24–month extension. Completers (n=116) at 24 months included 49 leflunomide, 21 placebo-sulfasalazine, and 46 sulfasalazine treated patients.

Study design. LEF= leflunomide; SSZ = sulfasalazine; PL = placebo; PL-SSZ = PL group switched to sulfasalazine; R = ACR20% responders; NR = ACR non-responders.
WITHDRAWALS
Table 2 summarises the withdrawals in each segment of the study. The proportion of total withdrawals was higher in the six month cohorts (leflunomide 37/133 (28%) v sulfasalazine 50/133 (38%)) than in the 24 month cohorts (leflunomide 11/60 (18%)v sulfasalazine 14/60 (23%)). Adverse events and lack of efficacy were the most common reasons for treatment withdrawals for both drugs. Withdrawals because of AEs were generally less with leflunomide than sulfasalazine throughout the study (14%v 19%, 3% v7%, and 10% v 15% at 6, 12, and 24 months, respectively).
Withdrawals
EFFICACY
Figure 2 shows changes in tender and swollen joint counts, calculated for each evaluation as the percentage change from baseline values. In the six month cohorts, both active groups showed significant improvements compared with placebo and these continued in the 12 and 24 month cohorts. The improvements in joint counts in the leflunomide and sulfasalazine groups were comparable at 6, 12, and 24 months (tender joint count 52%, 58%, 71% with leflunomide and 48%, 63%, 62% with sulfasalazine; swollen joint count 44%, 55%, 63% with leflunomide and 40%, 59%, 52% with sulfasalazine).

Mean changes (SD) in (A) tender and (B) swollen joint counts in 0–6 (LEF = 130, SSZ = 132, PL = 91), 0–12 (LEF = 78, SSZ = 74, PL-SSZ = 37), and 0–24 (LEF = 60, SSZ = 57, PL-SSZ = 25) patient cohorts at 6, 12, and 24 months. LEF = leflunomide; SSZ = sulfasalazine; PL = placebo; PL-SSZ = PL group switched to sulfasalazine. Baseline tender joint counts were: 0–6 month cohort (LEF = 18.8, SSZ = 16.7, PL = 16.3), 0–12 month cohort (LEF = 18.7, SSZ = 15.8, PL = 14.9), and 0–24 month (LEF = 18.4, SSZ = 15.7, PL = 14.1). Baseline swollen joint counts were: 0–6 month cohort (LEF = 16.2, SSZ = 15.3, PL = 15.8), 0–12 month cohort (LEF = 16.3, SSZ = 15.0, PL-SSZ = 14.4), and 0–24 month (LEF = 16.7, SSZ = 15.2, PL-SSZ = 13.9). Numbers in parentheses represent the percentage change from baseline. *p<0.0001 v PL.
Figure 3 shows changes from baseline in doctor and patient global scores. In the six month cohorts, significant improvements were observed with both active groups compared with placebo. Continued reductions in assessment scores were observed with leflunomide and sulfasalazine in 12 and 24 month cohorts. At 24 months, improvement in doctor (50% v 32%, p=0.03) and patient (46% v 30%, p<0.001) global assessments with leflunomide were significantly greater than with sulfasalazine.
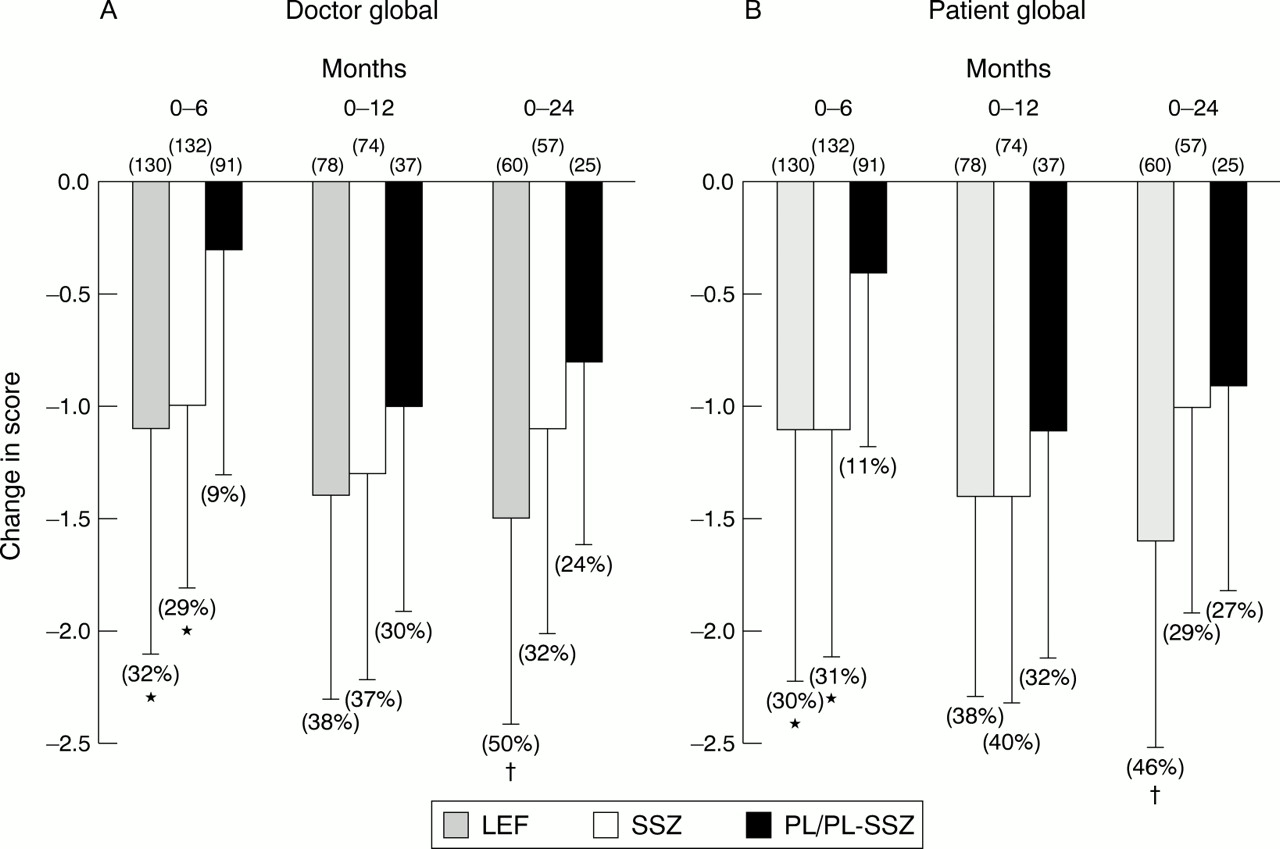
Mean changes (SD) in (A) patient and (B) doctor global scores in 0–6 (LEF = 130, SSZ = 132, PL = 91), 0–12 (LEF = 78, SSZ = 74, PL-SSZ= 37), and 0–24 (LEF = 60, SSZ = 57, PL-SSZ = 25) patient cohorts at 6, 12, and 24 months. LEF= leflunomide; SSZ = sulfasalazine; PL = placebo; PL-SSZ = PL group switched to sulfasalazine. Baseline doctor global scores were: 0–6 month cohort (LEF = 3.6, SSZ = 3.5, PL = 3.5), 0–12 month cohort (LEF = 3.7, SSZ = 3.5, PL-SSZ = 3.3), and 0–24 month (LEF = 3.6, SSZ = 3.4, PL-SSZ = 3.3). Baseline patient global scores were: 0–6 month cohort (LEF = 3.7, SSZ = 3.6, PL = 3.6), 0–12 month cohort (LEF = 3.7, SSZ = 3.6, PL-SSZ = 3.4), and 0–24 month (LEF = 3.7, SSZ = 3.5, PL-SSZ = 3.3). Numbers in parentheses represent the percentage improvement from baseline. * p<0.001 v PL; †p<0.001 v SSZ.
In the six month cohorts, changes from baseline in acute phase reactants, RF, duration of morning stiffness, pain intensity, and functional ability were significantly improved with both active groups compared with placebo (table 3). Continued reductions in all parameters were observed with leflunomide and sulfasalazine in 12 and 24 month cohorts. Changes in RF titres, morning stiffness, and pain intensity with leflunomide at 24 months reached ⩾60% improvement from baseline.
Changes in efficacy outcomes
In the six month cohorts, changes from baseline in Larsen scores were significantly improved with both active groups compared with placebo (table 3). Numerically greater reductions in Larsen scores were seen in 24 month cohorts with leflunomide than with sulfasalazine (−0.07v –0.03). These figures represent a 5% improvement from baseline with leflunomide in halting disease progression after 24 months of treatment. Sulfasalazine showed 2% improvement after treatment for 12 months (PL-SSZ group at 24 months) and 2% after 24 months of treatment. In the six month cohorts, changes from baseline in total erosive joint count were significantly lower with leflunomide and sulfasalazine (0.42 and 0.41, respectively) than with placebo (1.4, p<0.001). At 24 months, the leflunomide cohort showed a tendency towards fewer changes in erosive joint counts than the sulfasalazine cohort (−0.92 v 0.08).
We undertook a detailed analysis of the HAQ responses to evaluate the trend over time in functional ability, because leflunomide showed significant improvement in functional ability (in both mean HAQ scores and functional disability index) compared with placebo and sulfasalazine during the six month placebo controlled phase (table 3). Figure 4 shows overall mean changes in HAQ scores, and also depicts changes in ACR20% responders compared with non-responders. Improved HAQ scores in 6, 12, and 24 month leflunomide cohorts were observed in both non-responders (24%, 29%, 35%, respectivelyv sulfasalazine 8%, 10%, 27%) and ACR20% responders (63%, 62%, 66%, respectively vsulfasalazine 50%, 64%, 44%). These changes with leflunomide in non-responders throughout the study are in the range considered clinically significant25 ,26; even the six month non-responders in the leflunomide group showed a mean change of –0.29. In the sulfasalazine cohorts treated for up to one year, non-responders showed minimal improvement (<10%) in functional ability. At 24 months, improvement in HAQ scores (−0.65 v−0.36, p= 0.01) with leflunomide was significantly greater than with sulfasalazine. These data represent improvements of 56% and 45% in functional ability, respectively, with leflunomide and sulfasalazine in 24 month cohorts.

Changes in Health Assessment Questionnaire (HAQ) scores in 0–6 (LEF = 106, SSZ = 113), 0–12 (LEF = 66, SSZ = 62), and 0–24 (LEF = 51, SSZ = 45) patient cohorts at 6, 12, and 24 months. LEF= leflunomide; SSZ = sulfasalazine. Overall (O) HAQ scores as well as scores in ACR20% responders (R) and non-responders (N) are shown. *p<0.01, v SSZ.
The overall improvement seen in efficacy outcomes with leflunomide is reflected in the ACR response (fig 5). In the six month cohorts, significant improvements were seen with both active groups compared with placebo. Continued improvements in response were seen at 12 months. At 24 months, the ACR20% response rate with leflunomide was significantly greater than with sulfasalazine (82 %v 60%, p<0.01). Leflunomide was also significantly better than sulfasalazine at the more stringent 50% response rate (52% v 25%, p=0.04).

ACR20%, 50%, and 70% response rates in 0–6 (LEF = 130, SSZ = 132), 0–12 (LEF = 78, SSZ = 74), and 0–24 (LEF = 60, SSZ = 57) patient cohorts at 6, 12, and 24 months. LEF= leflunomide; SSZ = sulfasalazine. *p<0.05, v SSZ; †p<0.01, v SSZ.
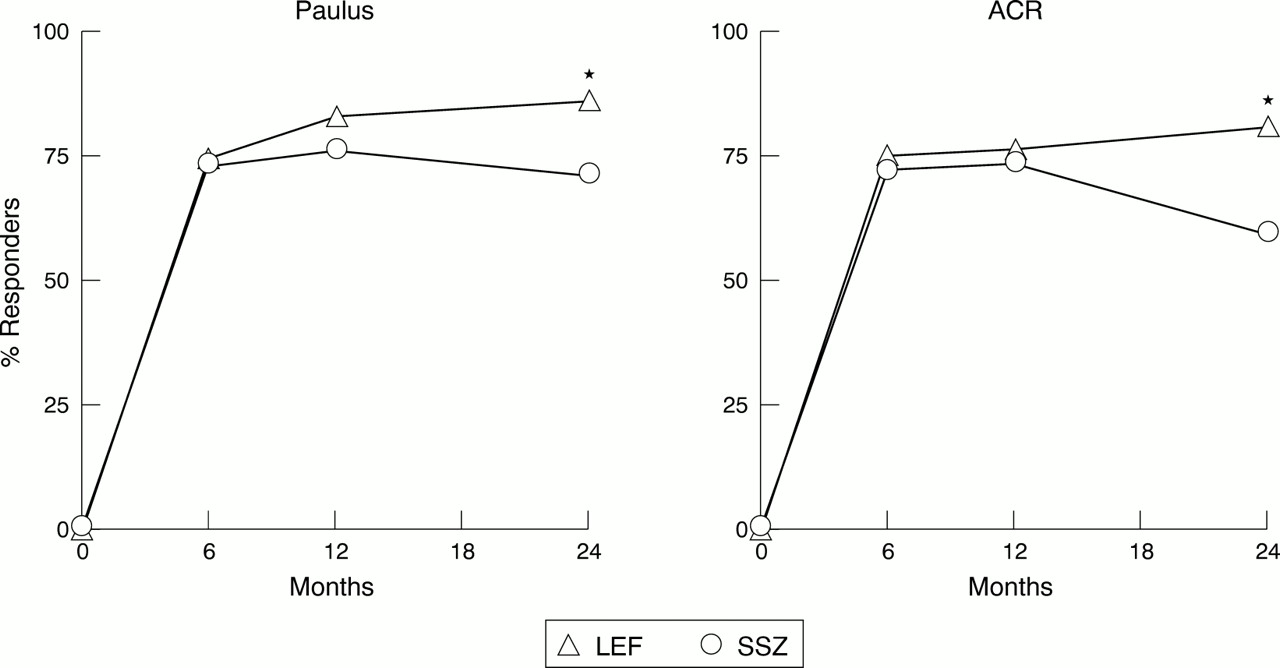
We also analysed completer responses to evaluate the trend over time in efficacy variables. As seen in fig 6, comparable results for overall response were seen using both Paulus and ACR20% criteria. Both leflunomide and sulfasalazine showed a rapid onset of action and comparable rates of response at six months. Response rates with leflunomide increased steadily over time. There was a slight decline with sulfasalazine response rates at 24 months, resulting in a significant difference versus leflunomide at 24 months.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Response rates (ACR20% and Paulus 20%) in completers (LEF = 60, SSZ = 57) from 0 to 24 months. LEF= leflunomide; SSZ = sulfasalazine. *p<0.05, v SSZ.
SAFETY
During the first six months, the most frequent drug related AEs in the leflunomide group were diarrhoea (leflunomide 17%, sulfasalazine 9%), nausea (leflunomide 10%, sulfasalazine 17%), and alopecia (leflunomide 8%, sulfasalazine 5%). Transiently raised liver function tests were seen in three leflunomide and five sulfasalazine treated patients. Two cases of reversible agranulocytosis in the sulfasalazine group were reported within six weeks of the start of treatment and required admission to hospital owing to infections. No unexpected AEs or late toxicity were seen with long term leflunomide treatment. Several AEs occurred less frequently with leflunomide in the second year, compared with the first six months (table 4). For example, the incidence of diarrhoea was reduced from 17% to 2%, alopecia, from 8% to 5%, and nausea, from 10% to 0%. Findings with sulfasalazine were comparable.
Adverse events at six months v 24 months. Data are shown as No (%)
During the initial six month study, hypertension, possibly related to the study drug, was reported in 2% of leflunomide, 1% of placebo, and 2% of sulfasalazine treated patients; most of these patients had pre-existing raised blood pressures. At 24 months, hypertension possibly related to the study drug was seen in 2% of leflunomide treated patients and in none of the sulfasalazine treated patients.
Levels of haemoglobin, packed cell volume, and erythrocyte counts increased, while white blood cell and platelet counts tended to normalise with leflunomide during the 6–24 month period. Packed cell volume and erythrocyte counts significantly decreased with sulfasalazine, while haemoglobin levels were unchanged. Increases in median values of liver function tests seen with leflunomide from baseline to 24 months (serum aspartate aminotransferase (AST) 17 U/l to 19 U/l or serum alanine aminotransferase (ALT) 14 U/l to 16 U/l, respectively) were small. Similar increases in liver function tests were seen with sulfasalazine (AST 17 U/l to 20 U/l or ALT 15 U/l to 16 U/l, at six and 24 months, respectively). No clinically relevant changes were found in other laboratory variables.
Four deaths occurred during the 12–24 month study period (two leflunomide, one sulfasalazine, one placebo-sulfasalazine); all were considered unrelated to the study drug. Three of the deaths were cardiogenic in origin: one leflunomide patient who died suddenly had a history of sinus tachycardia; the other leflunomide patient who died in cardiac arrest had developed deep vein thrombosis after 74 weeks of leflunomide treatment; and the placebo-sulfasalazine patient who died of acute myocardial infarction had coronary heart disease. The patient receiving sulfasalazine died of lung cancer. Cancer was reported in one patient receiving leflunomide (breast carcinoma) and six receiving sulfasalazine during the 12–24 month study period. Causal relation to the drug taken was implicated in only two of the sulfasalazine cases (non-Hodgkin's lymphoma with basal chronic lymphatic leukaemia and breast carcinoma with pseudopapillary clusters of the ductile epithelial cells).
Discussion
Previously reported six and 12 month data have shown that the efficacy and safety of leflunomide in patients with active RA was significantly better than placebo and comparable with sulfasalazine9 and methotrexate.10 However, longer follow up periods generally help in differentiating between DMARD activity. In the current communication we report that efficacy and safety with leflunomide are sustained for up to 24 months. The beneficial effects of leflunomide previously reported on function9 ,10 ,27 and disease progression9 ,28 are maintained for 24 months in patients who continue to receive treatment. Leflunomide was significantly better than sulfasalazine in improving global assessment scores, functional ability, and ACR response rates at 24 months.
Interpretation of data from a three stage design spanning a period of 24 months as reported is somewhat more limiting than data potentially obtained from a 24 month intention to treat analysis. This, coupled with the relatively low overall numbers of patients who continued in the 12 and 24 month extensions, means that these data should be viewed somewhat cautiously. However, continuance in the double blinded extensions after six months was voluntary, which, while not precluding selection of “good responders,” may not entirely represent a reflection of lack of efficacy. For example, of the 33 six month completers not participating in the 6–12 month extension, 7/16 in the leflunomide group, 3/7 in the sulfasalazine group, and 1/10 in the placebo group were ACR20% responders (fig 1). Moreover, the problems outlined above are those generally associated with long term follow up of placebo controlled studies. We believe that the data presented nevertheless demonstrate a clear trend of sustained efficacy over time in all variables, at least in a large subset of patients, that taken together with its benign safety profile attest to the effectiveness of a new DMARD, leflunomide, in RA. It is of some interest that those patients who continued to receive treatment for the full two year period were slightly more likely to be taking steroids; this observation, though tentative, is in keeping with the view that low dose steroid treatment is of considerable benefit to patients with RA.
A new DMARD such as leflunomide warrants long term follow up to establish its tolerability and to investigate potential delayed toxicity. Leflunomide is safe and well tolerated at doses of 20 mg over a two year period. Most toxicity occurred early with leflunomide and there were no unexpected side effects during the follow up. There was a decreased incidence of several AEs, such as diarrhoea, nausea, and alopecia, with continued treatment. A likely explanation of this finding is that some of this toxicity results from as yet unidentified genetic predispositions and invariably develops at an early stage during treatment. Withdrawals with both active groups were predominantly due to AEs rather than toxicity, although we recognise that it can be difficult to differentiate between these despite the best attempts to capture AEs in a formal manner. The total withdrawal rates during the placebo phase of the study were 28%, 45%, and 38%, respectively with leflunomide, placebo, and sulfasalazine. The incidence of total withdrawals with sulfasalazine reported in this study is in agreement with previously reported withdrawal rates of 33–38% at two years.29-31 A recent meta-analysis of randomised trials of sulfasalazine in RA reported total withdrawals of 40% with sulfasalazine.32 There was no clinically relevant haematological toxicity observed with leflunomide treatment. There were two cases of reversible agranulocytosis with sulfasalazine seen during the initial six month study period. Increases in levels of haemoglobin, packed cell volume, and erythrocyte counts, and decreases in platelet counts in the leflunomide group, suggest that leflunomide may improve anaemia induced by the chronic inflammation associated with active RA. Leflunomide shows a safety profile that is comparable with sulfasalazine, which is generally regarded as a DMARD with a relatively mild safety profile.33-35
One of the most striking differences in clinical efficacy reported between leflunomide and other DMARDs such as sulfasalazine and methotrexate is the improvement in HAQ scores.9 ,10 The current report substantiates this observation and shows that improvement in functional ability with leflunomide as compared with sulfasalazine persists for the duration of the follow up. It has been suggested that improvements of 36% above that of baseline or 18% above that of placebo constitute clinical benefit.21 At six months, leflunomide showed a 92% improvement above that of placebo, again exceeding the recommended 18% improvement above placebo. Changes in HAQ scores from baseline with leflunomide at 6, 12, and 24 months (44%, 50%, 59%, respectively) were also highly significant. Corresponding changes from baseline with sulfasalazine at 6, 12, and 24 months were 30%, 45%, and 40%, respectively. An Australian study of early RA noted no change in HAQ scores between sulfasalazine and placebo at six months.36 An open randomised trial in established RA reported no significant change in HAQ scores after 12 years of sulfasalazine treatment (baseline 2.13v 2.25 at 12 years).31
We further analysed the HAQ results found for leflunomide and sulfasalazine according to patient ACR responder status. The concept of patients with RA responding differently to a particular NSAID treatment is fairly well established.37-41 The HAQ data presented in the current paper suggest that a similar situation may exist with DMARD response. Substantial improvement was seen early in the treatment phase (six months) with leflunomide in both ACR responders and non-responders. With sulfasalazine, non-responders showed little or no change in functional ability during the first year of treatment. Early and sustained improvement in function with leflunomide in ACR responders and non-responders further delineates its effectiveness in RA.
A 12 year prospective study of patients with RA recently reported a strong relation between functional ability and disease activity scores (especially in pain intensity); however, correlations between HAQ andx ray scores were weak in the early phases of the study but became stronger at 12 years.42 Pain was reduced up to 59% with leflunomide, as were other parameters of disease activity; comparable changes were seen with sulfasalazine. More than 38% of patients enrolled in these leflunomide trials had disease duration of less than two years. The situation in early RA is more complex as in the initial stages of the disease, patients' HAQ scores are relatively high owing to their pain and synovitis. Devlin and his colleagues showed that suppressing the raised CRP in early RA was associated with a significant fall in HAQ scores.43 This finding suggests that part of the effect of leflunomide in reducing HAQ might be explained by a concomitant reduction in the acute phase response. Leflunomide demonstrated progressive reductions (51% to 60%) in CRP levels. Reduction in CRP at month 6 was significantly greater than with placebo (−23 mg/l v –20 mg/l, p<0.0001). There is also a relation between reduction in joint counts and improvements in the HAQ in early RA trials. Van Leeuwen and her colleagues found that in a three year prospective study of 149 patients with RA, HAQ scores were highly correlated with tender joint counts, and improvements in the HAQ were related to reduced numbers of tender joints.44 In our study tender joint count reductions were 52%, 58%, and 71% with leflunomide and 48%, 63%, and 62% with sulfasalazine at 6, 12, and 24 months.
Significant slowing of radiographic disease progression with leflunomide and sulfasalazine compared with placebo was seen at six months.9 In the current report, encompassing a period of two years, there was no further worsening of radiographically assessed articular damage, and leflunomide continued to slow disease progression, as seen in the mean changes from baseline in the Larsen score in the 0–24 month cohorts (leflunomide –0.07v sulfasalazine –0.03). Changes in erosive joint counts were significantly lower with leflunomide than placebo at six months (p=0.025); in 24 month completers the figure was –0.92, indicating improvement or halting of disease progression with leflunomide treatment. In a comparison with untreated controls, patients receiving sulfasalazine showed continuing deterioration at one year, but erosions were reported to slow down after two years.45 In a trial of sulfasalazine versus hydroxychloroquine in patients with early RA, significantly more radiographic damage was found with hydroxychloroquine at weeks 24 and 48,46 but the increase in total scores was not significantly different after 48 weeks. The high number of patients with early RA (<2 years) enrolled in these studies may account for the reported slowing of disease progression seen with leflunomide and sulfasalazine.
In summary, the data presented indicate that the clinical benefit of leflunomide is maintained at two years. Leflunomide is safe and well tolerated at doses of 20 mg with no unexpected AEs during the two year period. Improvement in functional ability, global assessments, and ACR response rates were significantly better with leflunomide than with sulfasalazine at two years. The pronounced early and sustained onset of improved function with leflunomide probably distinguishes it from other DMARDs. With the current trend of early intervention with DMARDs, leflunomide, which in clinical trial situations offers a good balance between efficacy and toxicity and a rapid onset of benefits on function and joint damage, may, with extended experience in routine clinical settings be a first choice when considering DMARD treatment.
The European Leflunomide Study Group
I Andreasson, Gothenburg, Sweden; P Andresen, Gråsten, Denmark; P Beck, Frederiksberg, Denmark; HA Bird, Leeds, UK; D Brackertz, Mainz, Germany; S Brighton, Pretoria, South Africa; H Bröll, Vienna, Austria; AK Clarke, Bath, Avon, UK; O Duke, Surrey, UK; W Graninger, Vienna, Austria; R Grigor, Auckland, New Zealand; B Hazleman, Cambridge, UK; P Jones, Rotorua, New Zealand; JR Kalden, Erlangen, Germany; AA Kalla, Capetown, South Africa; GH Kingsley, London, UK; R Kreuzeder, Vienna, Austria; TK Kvien, Oslo, Norway; GM Mody, Durban, South Africa; P Nash, Cotton Tree, Queensland, Australia; G Nuki, Edinburgh, UK; TG Palferman, Yeovil, Somerset, UK; M Pattrick, Crumpsall, Manchester, UK; P Pitt, Orpington, Kent, UK; PJ Prouse, Basingstoke, Hampshire, UK; F Rainer, Graz, Austria; B Rozman, PhD, Ljubljana, Slovenia; D Sahlberg, Oskarström, Sweden; M Schattenkirchner, Munich, Germany; DL Scott, London, UK; JS Smolen, Vienna, Austria; SF Sørensen, København, Denmark; M Tikly, Johannesburg, South Africa; LBA van de Putte, Nijmegen, Netherlands; R Westhovens, Pellenberg, Belgium; BD Williams, Cardiff, UK; R Williams, Hereford, UK.
References
Footnotes
↵* Members of the European Leflunomide Study Group are given in the .