Article Text

Abstract
Leflunomide is a selective inhibitor of de novo pyrimidine synthesis. In phase II and III clinical trials of active rheumatoid arthritis, leflunomide was shown to improve primary and secondary outcome measures with a satisfactory safety profile. The active metabolite of leflunomide, A77 1726, at low, therapeutically applicable doses, reversibly inhibits dihydroorotate dehydrogenase (DHODH), the rate limiting step in the de novo synthesis of pyrimidines. Unlike other cells, activated lymphocytes expand their pyrimidine pool by approximately eightfold during proliferation; purine pools are increased only twofold. To meet this demand, lymphocytes must use both salvage and de novo synthesis pathways. Thus the inhibition of DHODH by A77 1726 prevents lymphocytes from accumulating sufficient pyrimidines to support DNA synthesis. At higher doses, A77 1726 inhibits tyrosine kinases responsible for early T cell and B cell signalling in the G0/G1 phase of the cell cycle. Because the immunoregulatory effects of A77 1726 occur at doses that inhibit DHODH but not tyrosine kinases, the interruption of de novo pyrimidine synthesis may be the primary mode of action. Recent evidence suggests that the observed anti-inflammatory effects of A77 1726 may relate to its ability to suppress interleukin 1 and tumour necrosis factor α selectively over their inhibitors in T lymphocyte/monocyte contact activation. A77 1726 has also been shown to suppress the activation of nuclear factor κB, a potent mediator of inflammation when stimulated by inflammatory agents. Continuing research indicates that A77 1726 may downregulate the glycosylation of adhesion molecules, effectively reducing cell-cell contact activation during inflammation.
Statistics from Altmetric.com
Although the pathogenic mechanisms of rheumatoid arthritis (RA) remain elusive, advances in both molecular biology and clinical research have identified a unique orchestration of immune system cell subsets, cell surface markers, and soluble cell products that have a role in the process of inflammation associated with RA. Inflammation and subsequent degradation of the synovial tissue are initiated by the influx of lymphocytes (B cells, CD4+, and CD8+ T cells) into the synovial tissue. In the simplest model, CD4+ T lymphocytes are activated by antigens in the joint and stimulate plasma cells, mast cells, macrophages, and synovial fibroblasts to produce inflammatory mediators (tumour necrosis factor α (TNFα) and interleukin 1 (IL1)), which stimulate matrix degradation.1-3 Refining our knowledge of the immunoregulatory function of the various T cell subsets, as well as knowledge of the effects of macrophages and monocytes at the site of inflammation, may lead to the development of effective and targeted treatments for RA.
The T cell activation associated with RA has focused much attention on identifying drugs that regulate T cell progression through the cell cycle in an attempt to modulate proliferation.4 ,5 One such agent, leflunomide (N-(4-trifluoro-methylphenyl)- 5 - methylisoxazol - 4 - carbox- amide), is a low molecular weight (270), synthetic isoxazol derivative. Interest in leflunomide as an antirheumatic drug is due to its unique ability to regulate progression through the cell cycle by inhibiting de novo pyrimidine ribonucleotide biosynthesis.6-17
To be classified as a disease modifying antirheumatic drug (DMARD), a pharmaceutical agent must show efficacy and safety, as well as the ability to slow the progression of RA. Leflunomide (active metabolite A77 1726) is a unique DMARD with immunomodulatory properties. Effective treatment for RA requires a thorough knowledge of the mode of action of the therapeutic regimen chosen. In addition, with the current interest in combination treatment, the specific interaction between agents must be considered. Therefore, the purpose of this review is to describe the immunomodulatory actions of leflunomide in the treatment of RA at the cellular and molecular levels. In addition, the effects of combination treatment will be considered.
Leflunomide chemistry and metabolism
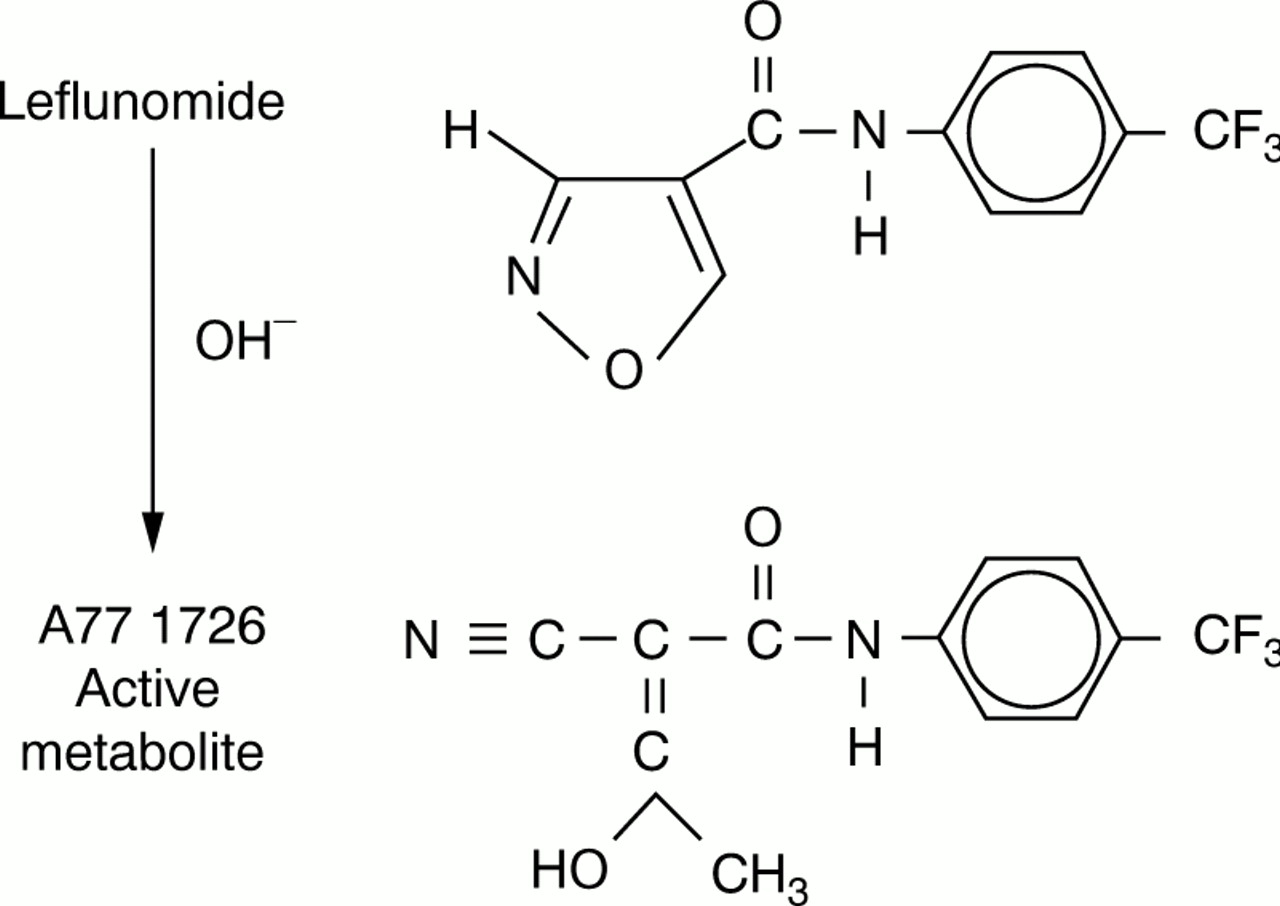
Leflunomide is a prodrug that is rapidly converted in the gastrointestinal tract and plasma to its active, open ring metabolite, the malononitrilamide, A77 1726 (2- cyano- 3-hydroxy-N-(4-trifluoromethylphenyl) butenamide) (fig 1). Structure-activity studies have shown how modifications to A77 1726 affect its immunoregulatory activity.10 ,18 ,19 In general, increased drug efficacy was observed when the 4-trifluoromethyl group of the aromatic ring was replaced by more lipophilic groups. Replacement of the aromatic ring by straight chain carbon groups decreased drug efficacy.

Chemical structure of leflunomide and its active metabolite A77 1726.
A phase II clinical trial with patients with RA showed that A77 1726 was highly bound to plasma protein (>99%) and had a half life of between 15 and 18 days. The total plasma clearance was 0.3 ml/kg/h.20 A somewhat shorter half life of about 11 days was reported in studies of healthy subjects.21 The majority of A77 1726 (60% to 70%) is eventually metabolised into trifluoromethylaniline-oxanilic acid and excreted in the urine.
A77 1726 regulates lymphocyte proliferation
A77 1726 has been shown to prevent skin, heart, and kidney graft rejection in animal models.22-24 Leflunomide is also effective in various experimental models of autoimmune diseases, including a spontaneous syndrome in MRL/lprmice that resembles human systemic lupus erythematosus,25 an anti-acetylcholine receptor induced model of myasthenia gravis in rats,26 and a T cell mediated allergic encephalomyelitis, which models human multiple sclerosis.27 More important for its role as an antirheumatic drug is the demonstration that leflunomide is highly effective in the treatment of animal models of both adjuvant28-31 and collagen induced arthritis.19
A77 1726 has been shown to regulate lymphocyte proliferation both in vitro7 9 12 13 32–36 and in vivo.14 ,31 ,36 ,37 In in vitro studies, A77 1726 is an equally effective immunoregulator of T cell proliferation when stimulated by cell surface receptor mediated mitogens or by mitogens, such as phorbol ester plus ionomycin, which bypass initial signal transduction events.9 ,12 ,34 T cell dependent B cell formation of autoantibodies, including IgA and IgG isotypes, is also inhibited by A77 1726.13 The dose of A77 1726 needed to modulate lymphocyte proliferation can vary greatly between species.12 ,34 In an in vitro study comparing proliferation of rat, mouse, and human lymphocytes, the mean 50% inhibitory concentrations (IC50 ) for A77 1726 were 0.09, 3.5, and 12.5 μmol/l, respectively.34 These IC50 values were similar for both receptor and non-receptor mediated (that is, phorbol ester plus ionomycin) stimulation.
Although the immunoregulatory properties of A77 1726 have been well documented, we have only recently begun to understand the molecular targets and biochemical mechanism(s) of action underlying these effects. Using flow cytometric analysis of mitogen stimulated T cells, Cherwinski et al showed that A77 1726 prevented the cells from entering the DNA replication phase (S phase) of the cell cycle34 (see also Siemaskoet al, 13 Langet al,37 and Herrmann M, Frangou CG, Kirschbaum B. Cell cycle control of the de novo pyrimidine synthesis inhibitor leflunomide through the p53 and p21WAF-1 pathways. Paper presented at the Association of Rheumatology Health Professionals 32nd National Scientific Meeting; 8–12 November 1997; Washington, DC). As illustrated in fig 2, A77 1726 may prevent cell progression into the S phase by acting at several sites in the G0 (resting) or G1 (growth) phases, or both. Currently, two specific mechanisms of action for A77 1726 have been identified: (a) inhibition of tyrosine kinases associated with the initial stage of signal transduction in G0, and (b) inhibition of de novo pyrimidine nucleotide biosynthesis in late G1. As will be described below, the inhibition of de novo pyrimidine nucleotide biosynthesis occurs at lower doses of A77 1726 than does the inhibition of tyrosine kinase activity and is considered the primary mechanism of action.

Cell cycle modulation. Some agents block signal transduction events in the resting G0 phase. Other agents interfere with ribonucleotide biosynthesis in the G1 phase. In either case, transition into the DNA replication phase, or S phase, of the cell cycle is blocked. NFAT = nuclear factor of activated T cells.
Inhibition of tyrosine kinase activity
Interference with the initial signal transduction events which play a part in T cell activation would prevent the transition of cells from the resting G0 phase to the G1 phase. Several processes involved in signal transduction, including the inhibition of tyrosine kinase activity, are affected by high doses of A77 1726. In Jurkat T cells, the activities of the Src related tyrosine kinases p56lck and p59fyn have been shown to be inhibited by A77 1726.33 Tyrosine phosphorylation of the chain of the CD3 complex, mediated by p59fyn, is thought to be critical for successful T cell antigen receptor/CD3 complex signalling. Likewise, p56lck mediated phosphorylation of the phospholipase C isozyme γ1 is important in the mobilisation of intracellular Ca2+. In another study A77 1726 was effective in inhibiting the levels of tyrosine phosphorylated proteins in mouse leukaemia cell line (LSTRA) cells, which overexpress p56lck. 11A77 1726 was also reported to suppress tyrosine phosphorylation in murine CTLL-4 cells stimulated by IL2.33 A similar inhibition of IL2 receptor (IL2R) associated tyrosine kinase activity has been seen in a staphylococcal nuclease-specific T cell line.38 More recently, Elder et al showed that A77 1726 inhibited the phosphorylation of the Jak1 and Jak3 tyrosine kinases, which are necessary for IL2R signalling.14
Other effects of A77 1726 on signal transduction have also been observed, but the data are more equivocal. Reductions in IL2 production, possibly related to inhibition of p56lck, have been reported in A77 1726 treated human T cells stimulated by anti-CD3 monoclonal antibodies.33 Cao et al showed that A77 1726 augments the immunosuppressive cytokine TGFβ1 (transforming growth factor β1), while simultaneously suppressing IL2.39 In contrast, another study failed to demonstrate an effect of A77 1726 in IL2 production.37 This latter observation is further supported by data demonstrating that exogenous IL2 is unable to antagonise the immunoregulatory effects of A77 172639 (Herrmann M, Frangou CG, Kirschbaum B. Cell cycle control of the de novo pyrimidine synthesis inhibitor leflunomide through the p53 and p21WAF-1 pathways. Paper presented at the Association of Rheumatology Health Professionals 32nd National Scientific Meeting; 8–12 November 1997; Washington, DC). Decreased IL2R expression has also been invoked as a potential mechanism of action of A77 1726.6 ,33 However, concentrations of A77 1726 which inhibit lymphocyte proliferation are not sufficient to reduce IL2R expression.9 ,34 ,37 Furthermore, many of the reported effects of A77 1726 on signal transduction might be secondary results of a primary mode of action (that is, dihydroorotate dehydrogenase inhibition).
Recent evidence suggests that A77 1726 is a potent inhibitor of nuclear factor-κB (NF-κB) activation.40 NF-κB is a transcriptional factor critical to the function of cells in the immune system, and it plays a part in inflammation. Manna and Aggarwal showed that treatment of human Jurkat T cells with A77 1726, at a concentration of 5–10 μmol/l, blocks TNF mediated activation of NF-κB.41 The effect was not constrained to TNF activation, as the stimulating effects of other inflammatory agents were also blocked. In contrast, tyrosine kinase activity was suppressed at a concentration of 150 μmol/l.12 A77 1726 targeted the degradation of the inhibitory protein IκBα, which is non-covalently associated with NF-κB in its inactive state in the cytoplasm. Inflammatory agents induce the phosphorylation dependent degradation of the inhibitory protein, which in turn unmasks the nuclear localisation signals on p65 and activates NF-κB. These observations indicate an additional biochemical mechanism for the immunomodulatory effects of A77 1726, as well as the observed anti-inflammatory actions.
Inhibition of tyrosine kinase is not the principal target of A77 1726
Several observations are incompatible with tyrosine kinase inhibition being the principal mechanism underlying the immunoregulatory action of A77 1726. A77 1726 is equally effective in inhibiting lymphocyte proliferation, whether it is stimulated through a cell surface receptor involving tyrosine kinases or by mechanisms that bypass these initial signal transduction events.13 ,34 It is also interesting to note that the ability of A77 1726 to inhibit proliferation was the same, whether it was added at the time of stimulation, eight hours later,34 or even up to 24 hours later.13 The drug's ability to affect proliferation, independently of time, questions the role of tyrosine kinase inhibition early in the signal transduction.
Comparison of the IC50 values for the regulation of lymphocyte proliferation versus inhibition of tyrosine kinase activity also suggests that early signalling events are not the principal target of A77 1726. A discrepancy between the IC50 values for A77 1726 mediated inhibition of proliferation and tyrosine kinase activity has been shown in the murine cytotoxic T cell line CTLL-4. The IC50 for inhibition of proliferation was 2 μmol/l, whereas that for tyrosine kinase mediated phosphorylation of Jak1 and Jak3 kinases was 50 μmol/l.14
Inhibition of de novo pyrimidine synthesis
During proliferation the pyrimidine nucleotide pool within lymphocytes must expand by approximately eightfold.42 In contrast, the purine nucleotide pool increases by only twofold.42 Although salvage pathways for obtaining pyrimidines exist, hereditary disorders of nucleotide metabolism suggest that highly proliferating cells such as lymphocytes need both de novo and salvage pathways to match the greatly increased demand for pyrimidine nucleotides during proliferation.42
Three independent reports appeared in 1995 suggesting that A77 1726 is a reversible inhibitor of pyrimidine nucleotide biosynthesis in vitro.6-8 Zielinski et al showed that exogenous application of the pyrimidine nucleotide, uridine, could reverse the antiproliferative action of A77 1726 in murine B and T cell lines studied in vitro.6In another study Cherwinski et al tested the ability of both pyrimidine and purine nucleotides to antagonise the antiproliferative actions of A77 1726 in several cell lines, including rat pheochromocytoma ACH2 cells, human Jurkat T cells, mouse B cell lymphoma A20–1.11 cells, and mouse B cell hybridoma 3DO-18.3 cells.7 The antiproliferative effect of 5 μM A77 1726 on all of these cell lines was completely blocked by 100 μM uridine. Cytidine, which can be converted to uridine by cytidine deaminase,43 also restored proliferation but with a reduced efficacy. The purine nucleotides, adenosine and guanosine, had no effect on the antiproliferative actions of A77 1726. Similar results were observed in mitogen stimulated normal rat, mouse, and human lymphocytes.
Using human Jurkat T cells, Cao et al found that uridine and cytidine (at concentrations of up to 4 μmol/l) were equally successful in reversing the immunoregulatory effects of A77 1726.9 This was true for concentrations of A77 1726 up to 24 μmol/l. At higher concentrations of A77 1726, uridine and cytidine were only partially successful in restoring proliferation. Presumably, tyrosine kinase activity was being inhibited at these higher concentrations of A77 1726. It is interesting to note that uridine was found to be effective in restoring proliferation when added to cell cultures up to 24 hours after the initial treatment with A77 1726.9
Subsequent studies have confirmed and extended the close association between the immunoregulatory actions of A77 1726 and altered pyrimidine nucleotide biosynthesis, both in vitro8 ,10-15 ,44 and in vivo.10 ,16 ,17 ,44 Changes in intracellular nucleotide pools caused by exposure to A77 1726 have also been measured.7 In human T lymphoblastoma CCRF.CEM cells, 10 μM A77 1726 decreased uridine triphosphate (UTP) and cytidine triphosphate (CTP) levels by 58% and 51%, respectively. In normal rat spleen cells stimulated with concanavalin A (Con A), 1 μM A77 1726 decreased UTP levels by 34% and CTP levels by 18%. At these doses of A77 1726 neither adenosine triphosphate (ATP) nor guanosine triphosphate (GTP) levels were affected. In IL2 stimulated murine T cells, 10 μM A77 1726 has been shown to decrease UTP levels by 95% and CTP levels by 85% without affecting either ATP or GTP levels.14 In the murine LSTRA cell line, A77 1726 selectively inhibits UTP and CTP synthesis with an IC50 of 10 μmol/l, which is similar to the IC50 for its antiproliferative activity (10–30 μmol/l).11 These results indicate that A77 1726 inhibits de novo pyrimidine synthesis.
A77 1726 is a non-competitive inhibitor of dihydroorotate dehydrogenase
A77 1726 could inhibit de novo pyrimidine nucleotide biosynthesis at several sites. There are six steps in the biosynthesis of uridine-5'-monophosphate (UMP) from ATP and glutamine. The enzymes which catalyse these steps are carbamyl phosphate synthetase II (CPSII), l-aspartate transcarboxylase (ATC- ase),l-dihydroorotase (DHOase), l-dihydroorotate dehydrogenase (DHODH), orotate phosphoribosyl transferase (OPRTase), and orotidine-5'-monophosphate decarboxylase (OMPDC). The first three enzymes in this pathway form a cytosolic multienzyme complex, CPSII-ATCase-DHOase. Because of their close association within this complex, enzymatic products are transferred efficiently from one active site to the next with minimum losses due to diffusion into the adjacent intracellular space. The fourth enzymatic step, catalysed by DHODH, occurs on the outer face of the inner mitochondrial membrane. Finally, the last two enzymes in the pathway form another cytosolic multienzyme complex, OPRTase-OMPDC.
The activities of all the enzymes in this pathway are allosterically controlled by the concentrations of both precursors and products.45 Two observations suggest, however, that the regulation of pyrimidine synthesis may be particularly sensitive to the catalytic step mediated by DHODH. Firstly, lymphocytes have fewer mitochondria than most cells.46 Secondly, both the precursor (dihydroorotate) and the product (orotate) of the DHODH mediated step must diffuse across the mitochondrial membrane. These two factors place DHODH at a strategic position to act as a key step in regulating the de novo synthesis of UMP. Consistent with this hypothesis, it has been shown that dihydroorotate accumulates in human T lymphoblastoid cells treated with A77 1726,8while exogenous orotate antagonises the effect of A77 1726.44
Other studies have shown directly the selective inhibition of DHODH by A77 1726. The effects of A77 1726 on the six different enzymes in the de novo UMP synthetic pathway were examined using a human T lymphoblastoma cell line.47 DHODH was shown to be reversibly inhibited with a Ki value of about 3 μmol/l. No other cytosolic enzymes in this biosynthetic pathway were affected. In another study the in vivo ability of a number of synthetic A77 1726 analogues to inhibit delayed-type hypersensitivity reactions in rats and mice was found to be closely correlated with their in vitro ability to inhibit DHODH activity.19 The inhibition of DHODH by A77 1726 has been confirmed in a number of subsequent in vitro studies.10 ,16 ,19 ,48 A consistent finding in all of these studies was that A77 1726 inhibited DHODH at drug concentrations similar to those resulting in its immunoregulatory effects, and at one to three orders of magnitude less than that needed for the inhibition of tyrosine kinases.
DHODH requires two substrates for catalysis. In addition to dihydroorotate, which is converted to orotate, ubiquinone is used as an electron acceptor. In recombinant human DHODH, it has been shown that A77 1726 inhibits the binding sites for both these substrates non-competitively.48 In a study analysing the kinetics of DHODH inhibition, it was found that A77 1726 acted as a competitive inhibitor of the ubiquinone binding site and a non-competitive inhibitor of the dihydroorotate binding site.48 These actions of A77 1726 distinguish it from another inhibitor of DHODH, brequinar sodium, which is a non-competitive inhibitor of both the ubiquinone and dihydroorotate binding sites.49 Figure 3summarises the effect of A77 1726 inhibition of DHODH on de novo synthesis of pyrimidine and on the subsequent activities of activated lymphocytes.

Effect of inhibition of de novo pyrimidine synthesis on various mechanisms of activated lymphocytes. (Adapted from Herrmann et al.49a)
A77 1726 decreases levels of rUMP leading to cell cycle arrest in late G1
In regulating the phosphorylation state of retinoblastoma protein (Rb), the cyclin D/cdk (cyclin dependent kinase) and E/cdk complexes serve as part of the G1 checkpoint, where cells with low ribonucleotide levels and/or damaged DNA are selectively prevented from progressing further into the cell cycle.50 Other components of the G1 checkpoint include a “sensor” to detect low ribonucleotide levels (that is, reduced rUMP (ribonucleotide uridine monophosphate) due to inhibition of DHODH through A77 1726) and damaged DNA, as well as an “effector” that inhibits the cyclin D/cdk and E/cdk complexes when such situations occur. The sensor function is accomplished by the tumour suppression protein, p53, whose proto-oncogene is also upregulated after a mitogenic stimulus.50-52
In dividing cells with adequate ribonucleotide levels, p53 is stabilised in the cytoplasm.50 In cells with low ribonucleotide levels, however, p53 is activated and translocates into the nucleus. If damaged DNA is detected, p53 induces the transcription of yet another regulatory gene, whose product is a 21 kilodalton protein, p21.53 The p21 protein acts as the effector for the G1 checkpoint. It inhibits the activity of the cyclin D/cdk and E/cdk complexes, leading to the dephosphorylation of Rb.53 As a result, Rb can rebind to the transcription factor E2F/DP-1, and cause the cell to become arrested in G1. Herrmann et al have shown in stimulated human peripheral T lymphocytes that both p53 and p21 become upregulated after exposure to A77 1726, resulting in the cells becoming arrested in late G1 (Herrmann M, Frangou CG, Kirschbaum B. Cell cycle control of the de novo pyrimidine synthesis inhibitor leflunomide through the p53 and p21WAF-1pathways. Paper presented at the Association of Rheumatology Health Professionals 32nd National Scientific Meeting; 8–12 November 1997; Washington, DC).
A77 1726 modulation of inflammation and metalloproteinase induction
At the site of inflammation in chronic inflammatory diseases, inflammatory cells and cells of the native tissue are in close proximity, which implies that a possible mechanism for cell communication is through direct cell-cell contact in addition to soluble factors. In a series of studies54-59 of cell-cell contact between T lymphocytes and monocytes, it has been shown that cell-cell contact induces the production of both matrix metalloproteinases (MMPs) and tissue inhibitor metalloproteinase-1 (TIMP-1). In addition, direct contact causes the upregulation of proinflammatory cytokines (IL1 and TNFα) and their inhibitors (IL1 receptor antagonist and TNF soluble receptor).60 It is postulated that an imbalance between MMPs and TIMPs and cytokines and their inhibitors may lead to the matrix destruction characteristic of chronic inflammation and, in particular, the matrix degradation associated with RA.
With these observations, Deage et alevaluated the effect of the anti-inflammatory agents, A77 1726 and dexamethasone, on the contact activation of the monocytic cell line THP-1.60 T lymphocytes were stimulated with phorbol myristate acetate and phytohaemagglutinin in the presence of both drugs, and it was noted that A77 1726 and dexamethasone inhibited the ability of stimulated T cells to activate the monocytic cells by 66%–97% and 43%–70%, respectively. It was found that A77 1726 tended to favour the inhibition of proinflammatory and matrix destructive factors over that of anti-inflammatory factors and MMP inhibitors (fig 4), thus potentially limiting matrix destruction. The exact mechanism for this effect remains to be determined. However, flow cytometry indicates that surface molecules (CD69 and CD11) of T lymphocytes that are, in part, involved in cell-cell contact signalling with monocytes, were not modulated by either drug, indicating an alternative mechanism.
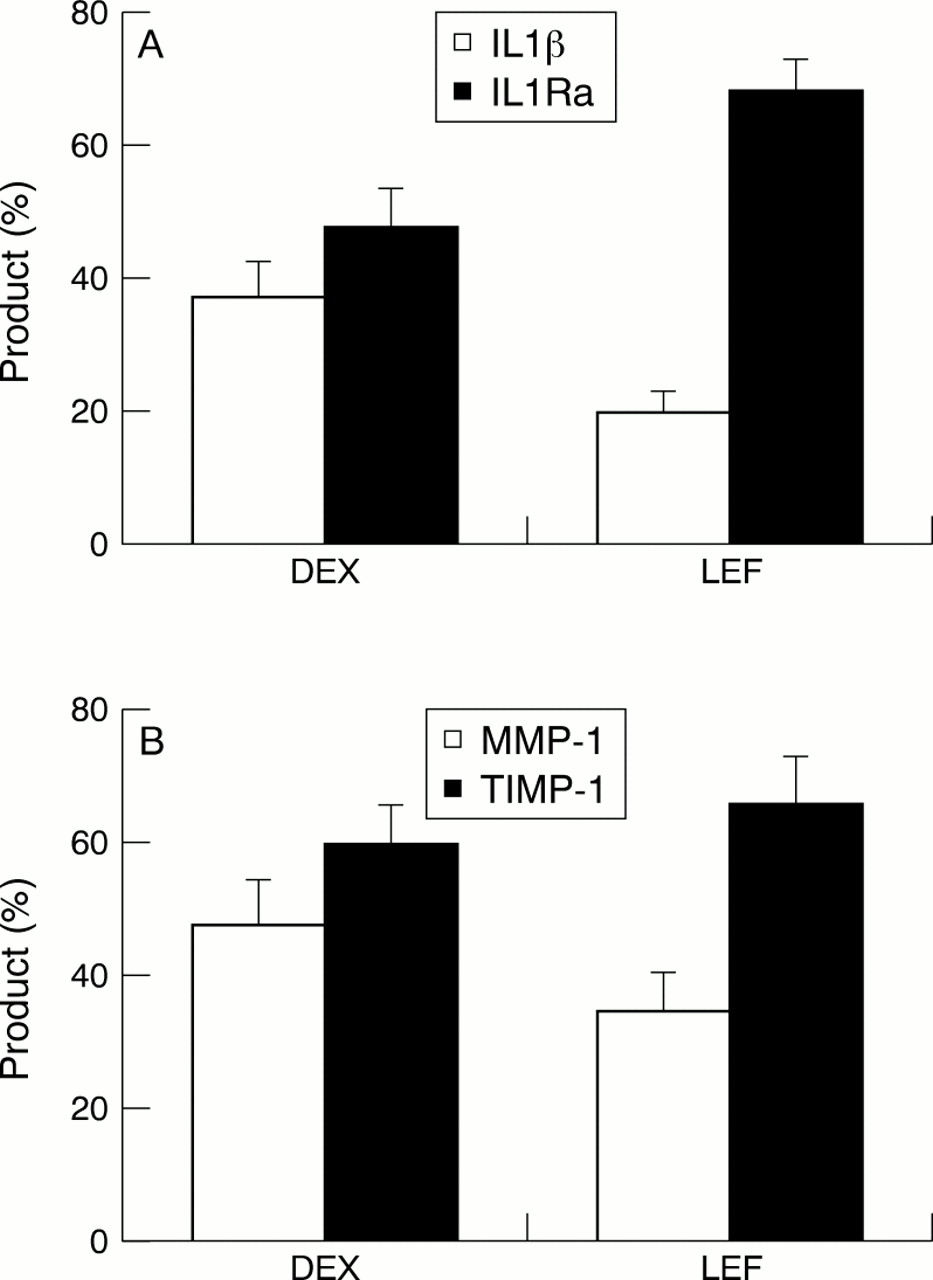
(A) Leflunomide (LEF) differentially inhibits the ability of stimulated T lymphocytes to activate THP-1 cells by direct cellular contact, favouring interleukin 1 receptor antagonist (IL1Ra) production, compared with dexamethasone (DEX). (B) Similarly, LEF differentially inhibits the ability of stimulated T lymphocytes to activate THP-1 cells by direct cellular contact, favouring the production of tissue inhibitor metalloproteinase-1. Concentrations of both LEF and DEX for figs 4A and B are 10−5mol/l.60
Preliminary clinical data on the expression of cell adhesion molecules (CAMs) and MMPs in synovial tissue from 39 patients in a prospective, double blind trial comparing leflunomide and methotrexate, indicate that both treatments resulted in comparable reductions in synovial inflammation and decreases in production of CAMs, and a decrease in the MMP/TIMP-1 ratio after four months of treatment (Kraan MC, Reece RJ, Barg EC, et al. An explorative study of the changes in synovial tissue in patients with RA after treatment with leflunomide or methotrexate. Paper presented at the 14th European League Against Rheumatism Congress; 6–11 June 1999; Glasgow, Scotland). Results of the study indicate that both methotrexate and A77 1726 showed comparable clinical efficacy in reducing inflammation within the synovial tissue.
In addition to providing strong evidence establishing that the primary mode of action of A77 1726 is the inhibition of DHODH, Rückemannet al also found that A77 1726 (100 μmol/l) significantly depleted ATP and GTP pools,61 thus strongly affecting ATP dependent enzymes, which modulate the immune system. By reducing ATP dependent pools of UTP, UDP-Glu (uridine diphosphoglucose), and CTP (fig 3), the subsequent expression of UDP sugars is inhibited, strongly influencing the glycosylation of adhesion molecules. Similarly, CTP lipids, which are essential for the incorporation of mannose and fructose into glycoproteins, as well as dolichol-linked intermediates through GDP sugar precursors, are suppressed (fig 3). Reduced GTP and GDP sugars would affect both lectin binding and cell surface topography, and as a result, would restrict mitogenic processes. These observations provide a mechanism for the anti-inflammatory and immunomodulatory actions of A77 1726.
Clinical overview of leflunomide: efficacy, safety, and combination therapy
The clinical efficacy and safety of leflunomide has been assessed in a one dose ranging phase II clinical trial,20 in two placebo controlled phase III clinical trials,62 ,63 and in one comparative phase III trial.64a
In both placebo controlled studies, clinical improvement, as defined by the American College of Rheumatology (ACR) 20% response criteria,64 was statistically significant for leflunomide (20 mg/day) compared with placebo. Leflunomide showed ACR 20% response rates comparable with those of sulfasalazine (2 g/day) and methotrexate (7.5–15 mg/week) in the placebo controlled clinical trials, and significant improvement in health related quality of life and functional ability compared with sulfasalazine and methotrexate.62 ,63 Leflunomide was also shown to reduce the rate of radiographically assessed disease progression significantly compared with the group receiving placebo and to produce comparable rates of progression compared with the groups receiving active treatment.62 ,63
The comparative two year trial of leflunomide versus methotrexate found both treatments comparable at two years in counts of tender joints and patient global assessment of disease activity. Radiographic assessments of disease progression at two years were also comparable.64a
The most common adverse events associated with leflunomide treatment were gastrointestinal, consisting primarily of diarrhoea, raised liver function tests, abdominal pain, and nausea/vomiting.20 ,62 ,63 The frequency and severity of the gastrointestinal complications were highest during the first two weeks of treatment with either leflunomide or methotrexate and declined thereafter.64a In addition, leflunomide has been associated with rash/allergic reactions, and reversible alopecia. Interestingly, there were no significant problems with anaemia, leucopenia, or thrombocytopenia, nor was there any increase in the incidence of infection compared with placebo. No opportunistic infections were noted.
Treatment with leflunomide may have a cytostatic effect, resulting from the inhibition of pyrimidine biosynthesis in tissues with a high rate of cellular turnover, such as those of the gastrointestinal tract. This speculative mechanism may also explain the incidence of reversible alopecia, as well as the lack of opportunistic infection, marrow toxicity, and mucositis65 seen in clinical trials of leflunomide.
The gastrointestinal adverse effects associated with high dose leflunomide (35 mg/kg/day) in a Lewis rat model of transplantation were diarrhoea and pathological changes of the small bowel and liver.66 These adverse effects were significantly reduced by uridine co-administration, which implies the mechanism leading to these side effects was related to DHODH dependent cell proliferation.
Early discussions of combination DMARD treatment67-69recognised the potential benefit in the treatment of patients unresponsive to traditional monotherapy, and, in particular, in the treatment of aggressive disease in early RA.68 ,69 Wilke and Clough highlight the need for detailed information about the pharmacology and modes of action for each of the combined drugs, and emphasise that DMARDs used in combination should have different modes of action.68 Furst concluded that effective combination treatment requires a thorough knowledge of the toxicity, modes of action, and pharmacokinetics of the combined DMARDs.70Because of its unique mechanism of action, the potential use of A77 1726 in combination treatment with DMARDs is promising.
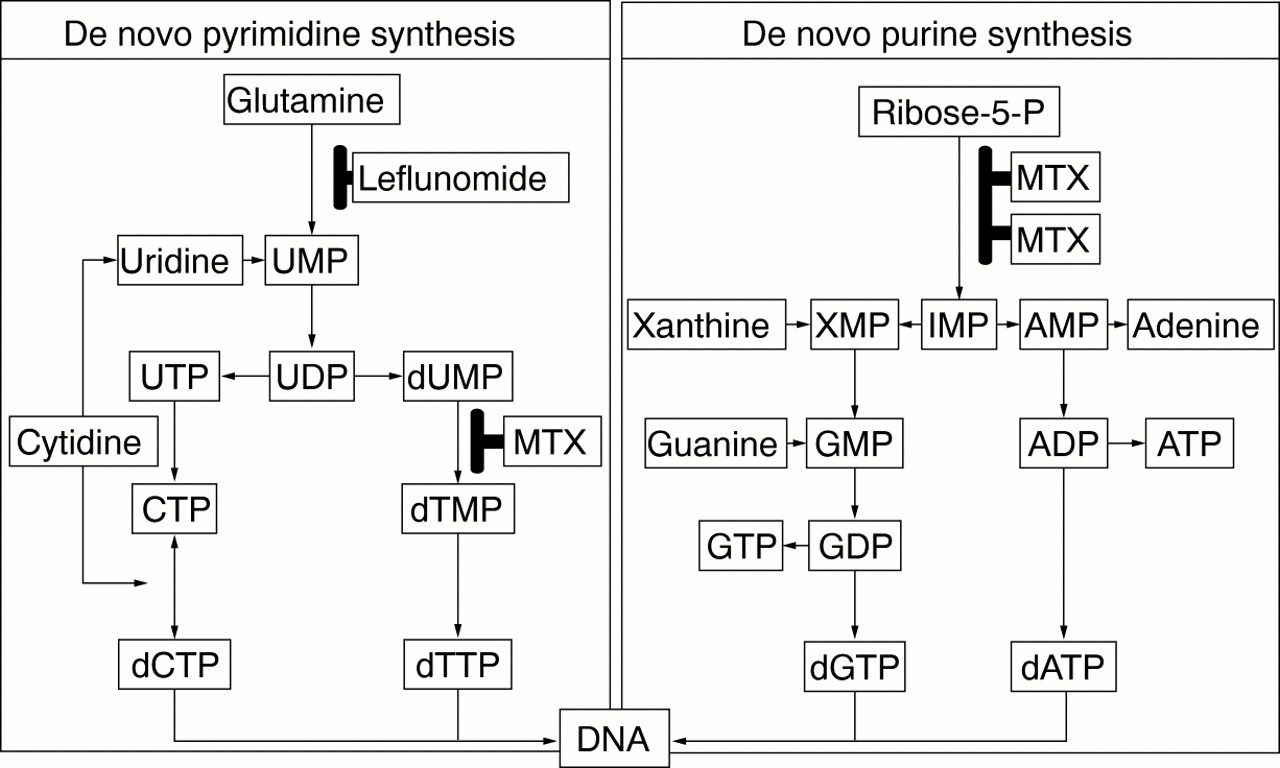
Methotrexate at the low doses used in the treatment of RA inhibits purine biosynthesis, and may have additional anti-inflammatory activity related to adenosine release. A77 1726, on the other hand, inhibits de novo pyrimidine biosynthesis and lymphocyte proliferation.71 Figure 5 summarises the unique modes of action of both leflunomide (active metabolite A77 1726) and methotrexate. The differing modes of action present a rationale for the combined treatment and may explain the synergistic effects of the two in combination.71 The combination of methotrexate and leflunomide has been proved to be effective for the treatment of patients with active RA who were refractory to treatment with methotrexate alone.64

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Comparison of the effect of leflunomide with that of methotrexate on nucleotide synthesis. MTX = methotrexate, dUMP = deoxyuridine monophosphate.
The clinical efficacy of combining A77 1726 with cyclosporin A has not been thoroughly investigated, but in vitro studies of the drug combination in animal models exhibit interesting complementary interactions. The rationale for combining A77 1726 with drugs such as cyclosporin A is that these early acting drugs may not prevent all autoimmune lymphocytes from entering G1. An additive interaction between A77 1726 and cyclosporin A in preventing T cell proliferation has already been shown in vitro,12 and in a canine renal transplant model.72 A77 1726, in combination with cyclosporin, resulted in the longest mean survival time, 68 days. It was also noted that a combination of A77 1726 and cyclosporin, both at suboptimal doses, effectively prolonged mean survival time by three times that for controls, and that the animals had normal renal function and weight. A similar effect was observed for antigen induced arthritis in rats, where a suboptimal dose of both drugs significantly inhibited chronic arthritis.73
The current prescribing information defines the recommended dose and administration of leflunomide. Owing to the long half life in patients with RA and the 24 hour dosing interval, a loading dose of 100 mg/day for 3 days is recommended, followed by a maintenance dose of 20 mg/day for the duration of treatment.74 If dosing at 20 mg/day is not well tolerated, the dose may be decreased to 10 mg/day. Liver enzyme levels should be monitored, and dose adjustments made, if necessary. Because of the long half life for the active metabolite, it is advised that during dose adjustment patients should be carefully monitored, as the levels of metabolite may take several weeks to reduce.74
Summary and conclusions
Leflunomide is a promising new antirheumatic drug with novel properties and mild side effects. The primary mechanism of action of this drug is the inhibition of de novo pyrimidine nucleotide biosynthesis. Unlike other proliferating cell types, lymphocytes cannot undergo cell division when the pathway for the de novo synthesis of pyrimidines is blocked. The ability of other cell types that undergo proliferation to use salvage pathways for acquiring pyrimidines may explain the relatively mild side effects of leflunomide.
New evidence shows that leflunomide inhibits the ability of T lymphocytes to stimulate monocytes through direct cell-cell contact in vitro. Leflunomide was shown to suppress preferentially the expression of proinflammatory molecules over their inhibitors, which implies a biochemical mechanism for the observed anti-inflammatory effects of leflunomide in the treatment of RA. Preliminary clinical data support this hypothesis. Synovial tissue biopsies of patients treated with leflunomide showed a decrease in the local production of CAMs and MMPs, contributing to the reported slowing of disease progression. The unique immunomodulatory effects of leflunomide show promise for the treatment of RA, alone and in combination treatment with other DMARDs.