Article Text

Abstract
Objectives Hyperplasia of synovial fibroblasts, infiltration with lymphocytes and tissue hypoxia are major characteristics of rheumatoid arthritis (RA). Extensive data support a key role for toll-like receptors (TLRs) in RA. Little is known regarding the impact of hypoxia on TLR-induced inflammation in RA. The aim of this study was to reveal the effects of hypoxia and its regulator, hypoxia-inducible factor-1α (HIF-1α), on the inflammatory response of RA synovial fibroblasts (RASF) to TLR ligands.
Methods Hypoxia was induced in RASF by incubation with Na2S2O4. TLR3 ligand polyIC, TLR2 ligand peptidoglycan, TLR4 ligand LPS and TLR9 ligand CpG were used to stimulate the cells. Effects of hypoxia on TLR-induced inflammatory mediators were determined by RT-PCR, qPCR and ELISA. Overexpression of HIF-1α as well as knocking-down its expression was used to reveal its fundamental role. RASF-induced inflammatory T cell expansion was determined by flow cytometry analysis of T helper (Th)1/Th17 cells, and IFN-γ/IL-17 production by ELISA after RASF/T cell coculture.
Results Hypoxia potentiated the expression of inflammatory cytokines, metalloproteinases and VEGF in RASF stimulated by different TLR ligands, especially polyIC, a synthetic mimic of dsRNA from viruses or apoptotic cells. HIF-1α played a fundamental role in this synergy. Moreover, HIF-1α overexpression enhanced RASF-mediated expansion of inflammatory Th1 and Th17 cells, leading to proinflammatory IFN-γ and IL-17 production.
Conclusions Our findings suggest that hypoxia and HIF-1α may function in conjunction with TLR-stimulated innate immune responses to drive inflammation in RA. This pathway may serve as a therapeutic target for the disease.
- Rheumatoid Arthritis
- Inflammation
- Fibroblasts
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) is a chronic systemic inflammatory disease characterised by hyperplasia of synovial fibroblasts and variable degrees of bone and cartilage erosion, leading to impairment of joint function. The pathogenesis remains to be fully elucidated.
Proinflammatory cytokines, such as interleukin (IL)-6, IL-8 and TNF-α, play dominant pathological roles in RA. Beside macrophages, RA synovial fibroblasts (RASF) are also an important source of these cytokines. RASF in the intimal lining were reported to be the primary source of IL-6.1 Aberrant T helper (Th) 1 cell response have long been considered to correlate with the pathogenesis of RA. Recent studies have further revealed the significance of Th17 cells in RA pathogenesis.2
Evidence has shown that RASF are not just passive responders in the rheumatoid joint, but can be potent participants in all aspects of RA pathogenesis.3 They can express different toll-like receptors (TLRs) as well as intracellular sensors of the innate immune system. Ligation of these receptors increases the expression of inflammatory cytokines, metalloproteinases (MMPs) and adhesion molecules in RASF, thus contributing to disease initiation, propagation and maintenance. TLR2, TLR3, TLR4 and TLR7 are particularly important in RA, with especially high expression levels.4 ,5
The microenvironmental conditions in the inflamed joint of RA patients are characterised by low partial oxygen pressure.6 ,7 Hypoxia-inducible factor 1α (HIF-1α), a key transcriptional factor in hypoxia, shows increased expression in RA.8 Hypoxia and HIF-1α could regulate many pathways in RA, such as inflammation, angiogenesis, migration and cell survival.9–12 Recent studies by Rius et al13 demonstrated that NF-κB links TLR signalling to the hypoxic response through transcriptional regulation of HIF-1α. Given the essential roles of TLRs in RA pathogenesis, it is important to understand the potential impact of hypoxia on TLR signalling-induced inflammation in RA.
In this study, we systematically explored the effects of hypoxia on the inflammatory response induced by TLR signalling in RASF, and further assessed the central role of HIF-1α as a mediator of exacerbations of inflammation.
Materials and methods
Patients and tissue specimens
Synovial tissue specimens used for culturing RASF were obtained from patients (n=6) during total knee replacement surgery or arthroscopy. All patients fulfilled the American College of Rheumatology 2009 criteria for RA. The study protocols and consent forms were approved by the Institutional Medical Ethics Review Board of Peking University People's Hospital.
RASF culture and treatment
RASF were cultured as previously described,3 and were used at passages 4–6. To induce hypoxia, 5×104 RASF per well in 6-well plates were pretreated with 1 mM Na2S2O4 (Sigma) for 2 h without serum as described.14 ,15 Then the cells were incubated with indicated stimuli for 4 and 8 h, respectively, as follows: polyinosinic-polycytidylic acid (polyIC) (25 µg/ml), peptidoglycan (10 µg/ml) and lipopolysaccharide (LPS) (100 ng/ml) from Sigma; and Type B pG oligodeoxynucleotides (CpG ODN) (10 µg/ml) from Invitrogen (Carlsbad, California, USA). The cells were harvested for RT-PCR and qPCR while the supernatants were collected for ELISA.
For gene silencing, TLR3-, MDA5- or HIF-1α-specific siRNA and the control scrambled siRNA (Santa Cruz, California, USA) were transfected into RASF according to the manufacturer's instructions, respectively. Briefly, 5×104 RASF were planted in 6-well plates per well overnight, then were transfected with 4 µl siRNA mentioned above using Transfection Reagent (Santa Cruz). After 24 h, the cells were tested for gene knocking-down efficiency and were treated as described above for qPCR and ELISA analyses. For HIF-1α overexpression, 5×104 RASF per well in 6-well plates were transfected with 2 µg HIF-1α plasmid. The cells were stimulated with polyIC 24 h later and subjected to qPCR and ELISA analyses.
RASF/T cell coculture assay
A quantity of 5×104 RASF were seeded in 6-well plates per well overnight, then were transfected with HIF-1α plasmid or vector as described above. After 24 h, 25 µg/ml polyIC was added and incubated for another 24 h. After that, RASF were washed with serum-free RPMI 1640 and cocultured with 2.5×105 allogeneic RA patient T cells (>95% purity) that were immunomagnetically separated by negative selection using RosetteSep Enrichment Cocktail (Stem Cell Technologies, Vancouver, British Columbia, Canada) at the ratio of 1:5 in the presence of anti-CD3 and anti-CD28 (3 µg/ml each). After 5 days, the T cells were harvested for flow cytometry while the supernatants were collected for ELISA.
RT-PCR and qPCR
Total RNA was extracted from cells using TRIzol reagent, treated with DNase, and then was examined by detecting A260/A280 as well as by agarose gel electrophoresis. An amount of 0.6 µg RNA was reverse transcribed into 20 µl cDNA and the resulting cDNA was diluted three times for PCR and qPCR (primers as in online supplementary tables S1 and S2). For qPCR, gene expression was quantified relative to the expression of the housekeeping gene GAPDH as well as β-actin, and normalised to control by standard 2-△△CT calculation.
ELISA assay
Commercially available ELISA kits used for measuring inflammatory cytokines and MMPs in the supernatants as well as HIF-1α in the nuclear cell lysates were as follows: IL-6, IL-8 and IFN-γ ELISA kits from Neobioscience Technology Co, Ltd (Beijing, China); IL-17 ELISA kit from QuantoBio Biotechnology Co, Ltd (Beijing, China); TNF-α ELISA kit from eBioscience (San Diego, California, USA); and MMP-3 and HIF-1α ELISA kits from R & D Systems (Minneapolis, Minnesota, USA).
Flow cytometry analysis
RA patient T cells were cocultured with RASF for 5 days as described above. At the end of coculture, the cells were stimulated with PMA (50 ng/ml) plus ionomycin (500 ng/ml) for 4–5 h in the presence of Brefeldin A (10 ug/ml, Becton Dickinson, San Diego, California, USA). Then, the cells were stained with PerCP-CY5.5-conjugated anti-CD4, fixed and permeabilised, followed by intracellular staining using FITC-conjugated anti-IFN-γ and Alexa Fluor647-conjugated anti-IL-17 (eBioscience). The percentage of positive cells was analysed on a FACS Arial II flow cytometer (Becton Dickinson).
Statistical analysis
SPSS V.17.0 (SPSS, Chicago, Illinois, USA) was used for statistical analysis. Differences between various groups were evaluated by Wilcoxon signed-rank test, and considered statistically significant when p was <0.05.
Results
Enhanced production of inflammatory cytokines in TLR3 agonist polyIC-stimulated RASF undergoing hypoxia
By measuring the level of HIF-1α, an indicator of hypoxia, we first assessed the effect of Na2S2O4 and polyIC on inducing hypoxia. Na2S2O4 treatment resulted in robust HIF-1α induction in RASF; polyIC slightly increased HIF-1α level. Additionally, polyIC, added together with Na2S2O4, resulted in an even higher HIF-1α expression (figure 1A, a–c).

Hypoxia enhanced polyIC-stimulated inflammatory cytokine production in rheumatoid arthritis synovial fibroblasts (RASF). (A) Induction of hypoxia-inducible factor-1α (HIF-1α) by pharmacological activation of hypoxia. RASF were pretreated with Na2S2O4 for 2 h, then stimulated with polyIC for another 4 h and were subjected to RT-PCR (a) and qPCR (b) analyses of HIF-1α. Also, 8 h after polyIC stimulation, the cells were harvested and the nuclear protein was extracted for ELISA analysis (c) of HIF-1α (Wilcoxon signed-rank test, *p < 0.005). (B–D) Enhanced IL-6, IL-8 and TNF-α production by hypoxia and polyIC. RASF were pretreated with Na2S2O4 for 2 h, and then were stimulated with polyIC for the indicated time. Generally, 4 h after stimulation, cells were harvested for RT-PCR (a) and qPCR (b) analyses, while 4 and 8 h after stimulation, cell culture supernatants were collected for ELISA (d). For kinetic analysis assays, RASF were stimulated with polyIC for 1, 4 and 8 h, respectively (c), and subjected to analyses (Wilcoxon signed-rank test, *p < 0.05). The results were presented as mean+SEM of six independent experiments.
Given the important role of inflammatory cytokines in RA, we next assessed the effect of hypoxia on polyIC-induced inflammatory cytokine production. Na2S2O4 treatment of RASF had a negligible effect on the basal levels of IL-6, but markedly enhanced polyIC-stimulated IL-6 expression (figure 1B, a–c), which was accompanied by elevated IL-6 protein levels in the supernatants (figure 1B, d). This synergy with hypoxia was also seen for polyIC-induced IL-8 (figure 1C) and TNF-α production (figure 1D). Interestingly, hypoxia also enhanced RASF production of IFN-β, a very important anti-inflammatory cytokine also resulting from polyIC stimulation (data not shown).
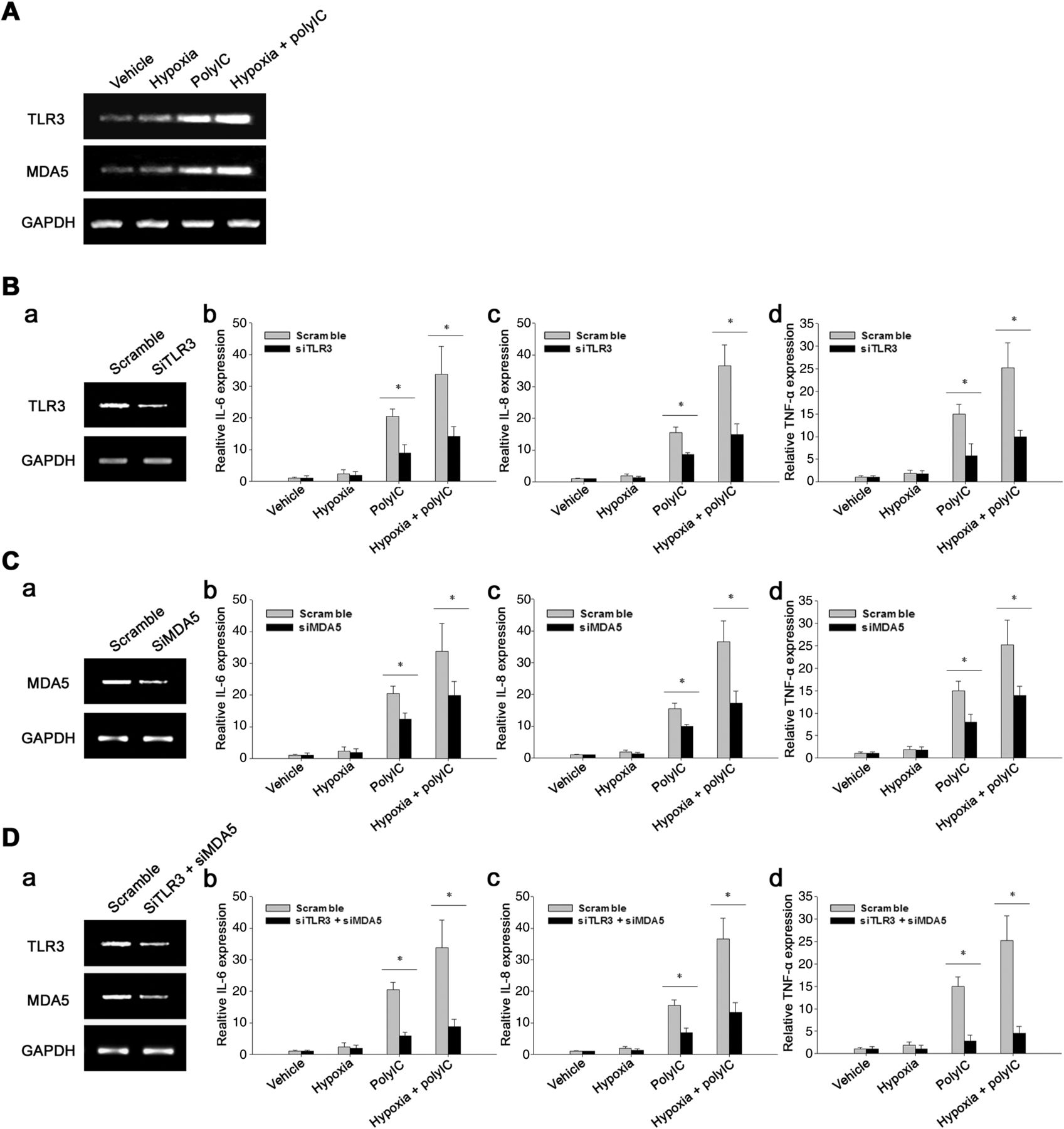
It is reported that the polyIC-induced inflammatory responses involve both TLR3 and MDA5,16 so we next examined the potential role of these two PRRs in the synergy of hypoxia and polyIC. PolyIC increased TLR3 and MDA5 expression in RASF, and Na2S2O4-induced hypoxia augmented this upregulation (figure 2A). Silencing the expression of either TLR3 (figure 2B) or MDA5 (figure 2C) resulted in diminished inflammatory cytokine expression, while knocking-down both these two receptors (figure 2D) abrogated the proinflammatory effect. These observations suggest that both TLR3 and MDA5 were involved in the synergistic action of hypoxia and polyIC.

Both toll-like receptor (TLR)3 and MDA5 were involved in hypoxia-enhanced inflammatory cytokine expression upon polyIC stimulation. (A) Rheumatoid arthritis synovial fibroblasts (RASF) were treated with hypoxia stressor Na2S2O4 alone, polyIC alone or their combination. TLR3 and MDA5 mRNA levels were examined by RT-PCR analysis. (B–D) Silencing TLR3 or MDA5 or both of them resulted in reduced inflammatory cytokine expression. (B) RASF were transfected with scramble siRNA or TLR3 siRNA for 24 h, and then were treated with Na2S2O4 for 2 h, followed by stimulation with polyIC for another 4 h. The TLR3 knocking-down efficiency was examined by RT-PCR (a). The IL-6 (b), IL-8 (c) and TNF-α (d) transcription were detected by qPCR analyses (Wilcoxon signed-rank test, *p < 0.05). Similar studies were performed for knocking-down MDA5 (C) as well as both TLR3 and MDA5 (D) expression (Wilcoxon signed-rank test, *p < 0.05). The results were presented as mean+SEM of four independent experiments.
Hypoxia synergised with TLR3 agonist polyIC to induce MMPs and VEGF production in RASF
MMP-dependent cellular invasion and VEGF-mediated angiogenesis are particularly important for RASF activity. So, next we assessed the effects of hypoxia on MMP and VEGF expression induced by polyIC. Na2S2O4-induced hypoxia potentiated MMP-1 expression in RASF, and further enhanced polyIC-stimulated expression, as measured by RT-PCR and qPCR (figure 3A,B). Similar results were also seen for MMP-3 (figure 3C,D), MMP-9 (figure 3E) and VEGF (figure 3F) production assessed by qPCR and ELISA.

RASF under hypoxia produced more metalloproteinases (MMPs) and VEGF upon the stimulation of polyIC. (A–E) Augmented production of MMPs in RASF by hypoxia and polyIC. Rheumatoid arthritis synovial fibroblasts (RASF) were pretreated with Na2S2O4 for 2 h and were stimulated with polyIC for another 4 h. Then the cells were harvested for RT-PCR analysis of MMP-1 expression (A). Accordingly, qPCR was also performed for detecting MMP-1 (B), MMP-3 (C) and MMP-9 (E) expression. Moreover, 8 h after polyIC stimulation, MMP-3 production was assessed by ELISA measurement of the protein levels (D) in the cell culture supernatants (Wilcoxon signed-rank test, *p < 0.05). (F) Hypoxia synergised with polyIC to induce more VEGF expression. RASF were treated as described above and subjected to qPCR analysis (Wilcoxon signed-rank test, *p < 0.05). The results were presented as mean+SEM of five independent experiments.
Effects of hypoxia on the inflammatory responses engaged by other TLR ligands
In addition to examining the effects of polyIC, we also assessed the impact of hypoxia on the inflammatory responses stimulated by other PAMP molecules. Na2S2O4 treatment of RASF strongly enhanced transcriptional activation of IL-6, IL-8 and TNF-α genes in response to TLR2 agonist peptidoglycan stimulation (figure 4A). Similar results were also seen for the TLR9 agonist CpG (figure 4B). In contrast, hypoxia compromised TLR4 agonist LPS-induced inflammatory cytokine production (figure 4C), which was consistent with previously reported results in macrophages.17 One potential mechanism is that hypoxia might diminish TLR4 expression in RASF, as reported by Ishida et al.18

Effects of hypoxia on inflammatory cytokine expression induced by different toll-like receptor (TLR) ligands. (A) Hypoxia enhanced inflammatory cytokine expression in TLR2 ligand peptidoglycan (PGN)-stimulated rheumatoid arthritis synovial fibroblasts (RASF). RASF cells were pretreated with Na2S2O4 for 2 h and stimulated with PGN for an additional 4 h. Cells were harvested for qPCR analyses of IL-6 (a), IL-8 (b) and TNF-α (c) expression (Wilcoxon signed-rank test, *p < 0.05). (B) Hypoxia enhanced inflammatory cytokine expression in TLR9 ligand CpG-stimulated RASF. RASF were pretreated with Na2S2O4, followed by CpG stimulation for 4 h and subjected to qPCR analyses of IL-6 (a), IL-8 (b) and TNF-α (c) mRNA levels (Wilcoxon signed-rank test, *p < 0.05). (C) Hypoxia dampened inflammatory cytokine expression in TLR4 ligand LPS-stimulated RASF. RASF were treated with Na2S2O4 and LPS as described above and were subjected to qPCR analyses of IL-6 (a), IL-8 (b) and TNF-α (c) expression (Wilcoxon signed-rank test, *p < 0.05). The results were presented as mean+SEM of four independent experiments.
Hypoxia regulator HIF-1α contributed to hypoxia-augmented inflammatory cytokine production
Given the important roles of HIF-1α in hypoxia-induced cellular signalling pathways as well as its linking function between innate immunity and hypoxia,13 we examined its potential involvement in hypoxia-amplified inflammatory responses. We first enforced the expression of HIF-1α in RASF by transfecting an HIF-1α plasmid, the presence of which was confirmed by qPCR and nuclear cell lysate ELISA (figure 5A, a,b). HIF-1α overexpression significantly enhanced IL-6, IL-8 and TNF-α expression induced by polyIC (figure 5A, c–e). ELISAs further validated this enhanced inflammatory response (figure 5A, f,g).

Hypoxia-inducible factor-1α (HIF-1α) contributed to hypoxia-induced augment of inflammatory cytokine production in response to polyIC stimulation. (A) HIF-1α overexpression synergised with polyIC to induce inflammatory cytokines. Rheumatoid arthritis synovial fibroblasts (RASF) were transfected with pcDNA3.1 or pcDNA3.1-HIF-1α for 24 h, and then were stimulated with polyIC. The HIF-1α mRNA level (a) as well as protein level (b) was first assayed to confirm the overexpression. Cells were harvested 4 h after stimulation for qPCR analyses of IL-6 (c), IL-8 (d) and TNF-α (e). Cell culture supernatants were collected 24 h later for measuring IL-6 (f) and IL-8 (g) protein levels (Wilcoxon signed-rank test, *p < 0.05, **p < 0.001). (B) Silencing HIF-1α abolished hypoxia-augmented inflammatory cytokine production induced by polyIC. RASF were transfected with scramble or HIF-1α siRNA. After 24 h, cells were pretreated with Na2S2O4, and then stimulated with polyIC for 4 and 8 h, respectively. HIF-1α knockdown efficiency was confirmed by RT-PCR (a), qPCR (b) and ELISA (c). The mRNA levels of IL-6 (d) and IL-8 (e) were assayed by qPCR, while the protein levels (f,g) were determined by ELISA (Wilcoxon signed-rank test, *p < 0.05). The results were presented as mean+SEM of five independent experiments.
To further confirm the contribution of HIF-1α to hypoxia-amplified inflammatory response, we silenced HIF-1α by specific siRNA. The HIF-1α knockdown efficiency was confirmed by RT-PCR, qPCR and ELISA (figure 5B, a–c). As shown in figure 5B, d,e, when HIF-1α was knocked down, hypoxia-enhanced IL-6 and IL-8 expression in polyIC-stimulated RASF was abrogated. These findings were further confirmed by ELISA (figure 5B, f,g). These results suggest that HIF-1α, at least in part, coupled hypoxia to inflammatory cytokine production in the context of polyIC stimulation.
HIF-1α overexpression enhanced RASF-mediated expansion of inflammatory T cells
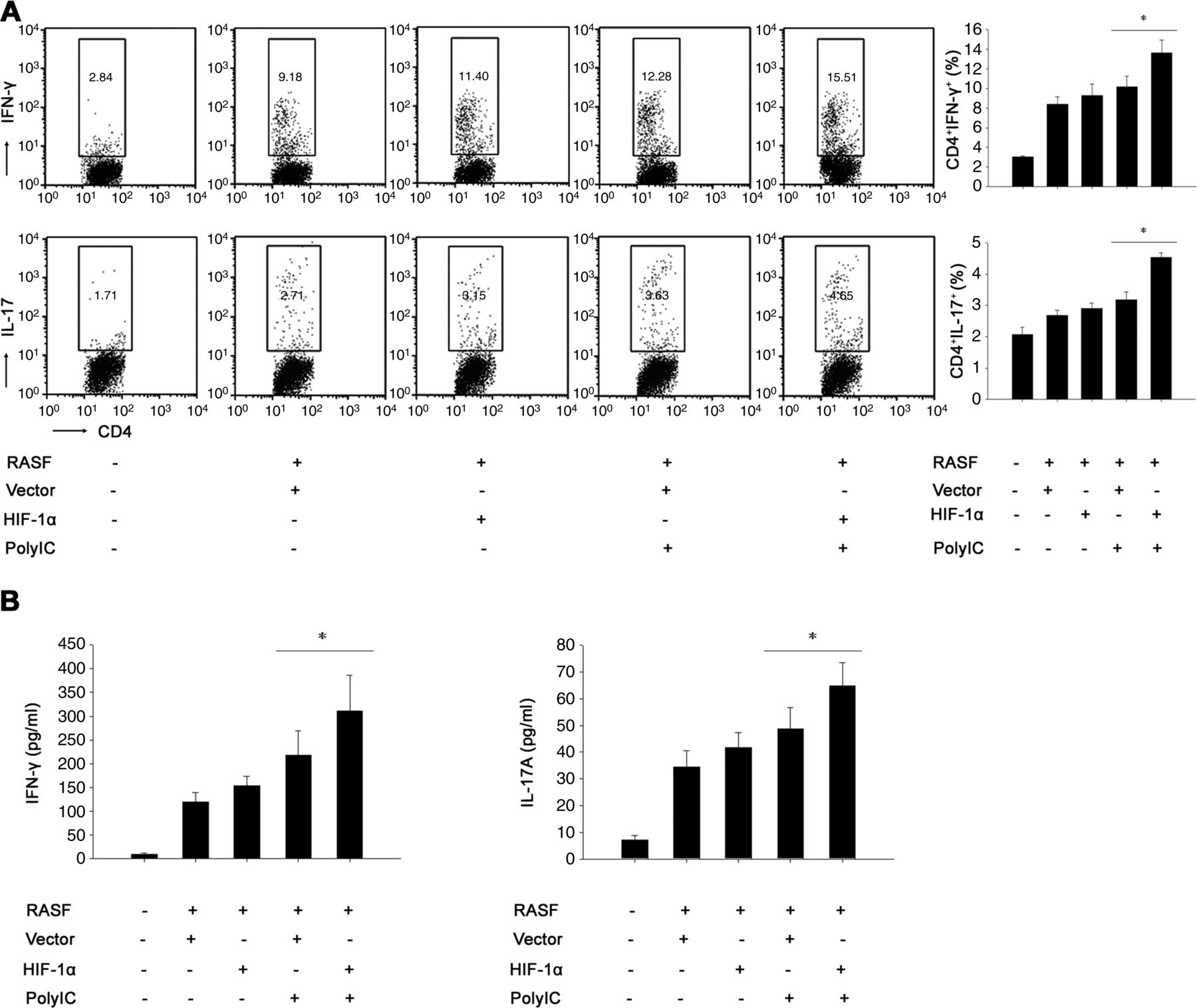
RASF could inhibit the apoptosis of T cells through cell–cell interaction and/or elaboration of soluble factors, which elicits spontaneous proliferation of inflammatory T cells. The increased inflammatory T cells then in turn induce more robust RASF activation, forming a RASF/T cell regulatory circuit that perpetuates the inflammatory process in RA.3 So, we next determined the impact of HIF-1α overexpression on RASF-mediated expansion of inflammatory T cells. RASF were transfected with HIF-1α construct or control vector and then were treated with polyIC, followed by coculture with RA patient T cells. Flow cytometry showed that HIF-1α overexpression amplified RASF-mediated expansion of inflammatory Th1 and Th17 cells (figure 6A), resulting in a shift toward a proinflammatory state. Moreover, as shown in figure 6B, higher levels of IFN-γ and IL-17 were seen in the supernatants of T cells cocultured with RASF overexpressing HIF-1α after polyIC stimulation. All these suggest that HIF-1α overexpression enhanced RASF-mediated expansion of inflammatory T cells.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Enhanced expansion of inflammatory T cells mediated by rheumatoid arthritis synovial fibroblasts (RASF) overexpressing hypoxia-inducible factor-1α (HIF-1α) upon the stimulation of polyIC. (A) HIF-1α overexpression amplified the expansion of inflammatory T helper (Th)1 and Th17 cells mediated by RASF. RASF were transfected with pcDNA3.1 or pcDNA3.1-HIF-1α for 24 h, and then were stimulated with polyIC for another 24 h. After that, the cells were cocultured with allogeneic T cells isolated from RA patients in the presence of anti-CD3 and anti-CD28. After 5 days, the T cells were harvested for flow cytometry analysis of the percentage of Th1 and Th17 cells (Wilcoxon signed-rank test, *p < 0.05). (B) This HIF-1α overexpression induced a shift toward the proinflammatory state. RASF-T cell coculture was performed as described above, and the cell culture supernatants were collected for ELISA analyses of IFN-γ and IL-17 levels (Wilcoxon signed-rank test, *p < 0.05). The results were presented as mean+SEM of four independent experiments.
Discussion
The innate immunity mediated by TLRs in RASF plays a crucial role in the initiation and in the perpetuation of RA. The excessive synovial expansion outstrips the oxygen supply, leading to synovial hypoxia. This study is the first to show that hypoxia and HIF-1α potentiated the production of the inflammatory cytokines, MMPs and VEGF in RASF induced by TLR signalling, especially polyIC-mediated signalling. This synergy further enhanced RASF-mediated expansion of inflammatory T cells, perpetuating the inflammation in RA.
Infection has long been speculated to be an underlying factor in RA pathogenesis. The presence of peptidoglycan, bacterial and viral DNA was shown in RA joints in early studies, implicating PAMP activation of TLRs as a potential driver of the disease.19 Moreover, endogenous TLR ligands such as HSPs, HMGB1 and double stranded RNA have been demonstrated in inflamed RA joints, implying a role for DAMP activation of TLRs in RA pathogenesis.20 In this study, we showed that hypoxia synergised with peptidoglycan, CpG (a synthetic mimic of DNA from bacteria or apoptotic cells) and polyIC (a synthetic mimic of RNA from virus or apoptotic cells) to induce more robust inflammatory mediator production. Since the microenvironment of the inflamed RA patient joints is hypoxic, our results suggest that during bacterial or viral infection or after exogenous danger molecule encounters, synovial fibroblasts become more active to produce inflammatory mediators, thus exacerbating RA severity.
Hypoxia is an evolutionarily ancient stress response triggered by low ambient oxygen and controlled by HIF-1, especially HIF-1α. As another evolutionary ancient stress response, the innate immunity mediated mainly by TLRs is regulated by several transcriptional factors, among which NF-κB plays a central role. Many studies have revealed the correlation between the two responses. Rius et al13 demonstrated that NF-κB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1α in macrophages. This could explain the synergistic effects of hypoxia on TLR signalling-induced production of inflammatory cytokines as well as MMPs and VEGF since their production is controlled by NF-κB. Nevertheless, the detailed mechanism, for example, whether hypoxia facilitates the accessibility of NF-κB to the promoters of these inflammatory mediators, remains to be further explored.
As the key regulator of hypoxia, HIF-1α plays an important role in the pathogenesis of RA. Upregulated expression of HIF-1α was shown in RA patient synovial tissues.8 It could mediate the recruitment of monocytes, T and B lymphocytes in the rheumatoid synovium.9 Besides, it promotes MMP and VEGF expression in RASF, inducing cartilage destruction and angiogenesis.11 ,12 In our study, enforced HIF-1α expression in RASF recapitulated the effects of hypoxia on polyIC-triggered inflammatory response, while knocking-down its expression abrogated the synergy between hypoxia and polyIC. This further elucidated the potential bridging role of HIF-1α in hypoxia and innate immunity. Moreover, in the current study, we showed that overexpression of HIF-1α enhanced RASF-mediated expansion of inflammatory Th1 and Th17 cells, inducing a shift toward the proinflammatory state. Upregulated expression of thrombospondin 1 and IL-15 on RASF that mediate the cell–cell contact between RASF and T cells, as well as increased soluble factors secreted by RASF, particularly IL-6 and TGF-β, should contribute to this expansion.21 In addition, HIF-1α could directly enhance Th17 development and attenuate Treg development in mice. Mice with HIF-1α-deficient T cells are resistant to Th17-dependent experimental autoimmune encephalitis.22 Possibly, HIF-1α also controls Th17/Treg balance in human with the same pattern, thus contributing to RA pathogenesis, which deserves to be further studied.
In summary, we have shown that hypoxia potentiated TLR signalling-induced inflammation in RA via HIF-1α. This improves our understanding about the pathogenesis of RA, and further suggests HIF-1α as a new therapeutic target for overcoming the persistent and chronic inflammatory disease.
Acknowledgments
We gratefully thank Dr Taiping Shi (Chinese National Human Genome Center, Beijing, China) for kindly providing the HIF-1α plasmid. This work was supported by grants from 973 program of China (2010CB529100) and the Natural Science Foundation of China (81120108020 and 81030057) as well as by Peking University People's Hospital Research and Development Funds (RDB2012–04).
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online table 1
- Data supplement 2 - Online table 2
Footnotes
Handling editor Tore K Kvien
-
Contributors FH and ZL designed the research. FH performed the research and analysed the data. FH, PC, XQ and ZL wrote the manuscript. RM, JZ, LS, YL, XL, WS, GL, ML and YS helped with sample collection and data analysis.
-
Funding 973 program of China; the Natural Science Foundation of China; Peking University People's Hospital.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval Institutional Medical Ethics Review Board of Peking University People's Hospital, Beijing, China.
-
Provenance and peer review Not commissioned; externally peer reviewed.