Article Text

Abstract
Objective To assess the safety and efficacy of secukinumab, a fully human monoclonal anti-interleukin-17A antibody, in patients with rheumatoid arthritis (RA).
Methods Patients (n=237) with inadequate response to methotrexate were randomly assigned to receive monthly subcutaneous injections of secukinumab 25 mg, 75 mg, 150 mg, 300 mg or placebo. The primary endpoint was the American College of Rheumatology 20% response (ACR20) at week 16.
Results Demographics and baseline characteristics were comparable across all treatment groups. The primary efficacy endpoint was not achieved: the proportion of ACR20 responders at week 16 with secukinumab 25–300 mg was 36.0–53.7% versus placebo (34%). Disease activity score in 28 joints (DAS28)–C-reactive protein (CRP) was a secondary endpoint and clinically relevant decreases with secukinumab 75–300 mg were reported versus placebo. Serum high sensitivity CRP levels at week 16 were significantly reduced with secukinumab 75 mg, 150 mg and 300 mg doses versus placebo. The safety profile of secukinumab was consistent with that seen with other biological agents. Most adverse events (AE) were mild to moderate in severity. Infections were slightly more frequent with secukinumab than placebo. Six serious AE were reported: secukinumab 75 mg (one), secukinumab 300 mg (four) and placebo (one).
Conclusions ACR20 response rates differed between secukinumab 75 mg, 150 mg and 300 mg doses and placebo; however, the primary efficacy endpoint was not achieved. Greater decreases in DAS28 were observed with secukinumab 75 mg, 150 mg and 300 mg than placebo. There were no unexpected safety signals and no specific organ-related toxicities. Further trials with secukinumab in the treatment of RA are warranted.
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) is a chronic, inflammatory, systemic autoimmune disease of unknown aetiology. It is characterised by symmetrical synovitis leading to cartilage damage and joint destruction, and can be complicated by numerous extra-articular manifestations. It is generally a progressive disease with functional decline, significant morbidity and premature mortality.1 ,2
Current treatment of RA with disease-modifying antirheumatic drugs (DMARD), such as methotrexate, is inadequate in many patients because these drugs only partly control established disease. Targeted therapies have been introduced for the treatment of RA and other immune-mediated diseases, and are generally prescribed together with methotrexate.3 A number of biological drugs are now approved for use in RA and target tumour necrosis factor (TNF)α, interleukin (IL) 1 receptor, IL-6 receptor, costimulation through CTLA-4 and B cells through depletion with anti-CD20. In general, anti-TNF agents are the first line biological agents used in DMARD failure patients, but 30–40% of patients fail to respond to TNFα antagonists and the majority of those who respond initially do not achieve complete remission or lose response over time.4 ,5 There remains an unmet need for patients with RA, thus it is warranted to investigate further new targets and develop drugs with alternative mechanisms of action for RA treatment.
The discovery of IL-17 and the T-helper (Th) 17 lymphocyte has been a major step in the understanding of erosive arthritis.6 IL-17A, commonly referred to as IL-17, is a key member of the IL-17 family, of which there are six, A–F.7
The Th17 pathway is thought to be an important mediator of autoimmunity that is distinct from previously described T-cell pathways in disease (eg, Th1/Th2). Emerging evidence demonstrates that IL-17 can also be synthesised by myeloid lineage cells in inflammatory arthritis tissues. Functionally, IL-17A exerts potent proinflammatory and joint-destructive activities such as stimulation of IL-1 and TNF production from human macrophages,8 induction of IL-6 and IL-8 secretion in synovial fibroblasts9 and upregulation of RANKL, which in turn promotes osteoclastogenesis10 and thereby contributes to bone erosions. In addition, IL-17A promotes cartilage degradation by inducing metalloproteinases and proteoglycan depletion.11 ,12 Inflamed tissues contain abundant IL-17A producing cells, and circulating Th17 cell numbers and synovial fluid IL-17A strongly correlate with disease activity in RA.13 ,14
Secukinumab (Novartis Pharma AG, Basel, Switzerland) is a high-affinity, fully human anti-IL-17A monoclonal antibody that selectively binds to and neutralises IL-17A, both in vivo and in vitro. Neutralisation of IL-17A with secukinumab thus affords a novel opportunity to intervene in a pro-inflammatory pathway elicited by a cytokine derived from both adaptive and innate immune sources in the autoimmune arthritides such as RA.
Neutralisation of IL-17A has compelling preclinical6 ,15–18 and clinical target validation. In a proof of concept study treatment with secukinumab resulted in significant improvement of signs and symptoms in patients with RA relative to placebo, providing evidence that the neutralisation of IL-17A with secukinumab in combination with methotrexate could be effective in patients with active RA (CAIN457A2101).19
Herein, we report the interim analysis results of efficacy up to week 16 and safety up to week 20 of a 52-week phase II study designed to assess the efficacy and safety of different doses of secukinumab in patients with active RA despite treatment with stable doses of methotrexate.
Methods
Patients
Patients aged 18 years or older, with a diagnosis of active RA according to the revised 1987 American College of Rheumatology (ACR) criteria, for 3 months or more were enrolled in this study. Active disease was defined by six or more out of 28 tender joints and six or more out of 28 swollen joints, and acute phase reactant (high sensitivity C-reactive protein (hsCRP) ≥10 mg/l or erythrocyte sedimentation rate (ESR) ≥28 mm/first hour).20 Allowed concomitant medications were stable doses of methotrexate (≥7.5 mg/week to ≤25 mg/week for ≥3 months), corticosteroids (≤10 mg/day prednisone or equivalent) and non-steroidal anti-inflammatory drugs for at least 4 weeks at the time of randomisation. Patients who had failed DMARD and/or biological agents were allowed to participate in the study after an appropriate wash-out period before randomisation. Patients with RA functional status class IV according to the revised ACR 1991 criteria21 were excluded. Women of childbearing potential were required to use appropriate contraception, as defined in the protocol. The use of prednisone greater than 10 mg/day, high potency opioid analgesics or any therapy by intra-articular injections required for treatment of acute RA flare within 4 weeks before randomisation was not allowed. Patients with ongoing other rheumatic diseases other than RA (including but not limited to primary fibromyalgia, ankylosing spondylitis, psoriatic arthritis, and reactive arthritis), any active or recent infections, or a history of malignancy were also excluded.
Study design
This was a multicentre, dose-finding, double-blind, randomised, placebo controlled study conducted in 54 centres in 11 countries: Belgium, Czech Republic, Germany, Hungary, Japan, Korea, Poland, Russia, Slovakia, Taiwan and USA. Adult RA patients (N=237) on methotrexate were randomly assigned equally to receive monthly subcutaneous injections of secukinumab 25 mg, 75 mg, 150 mg, 300 mg or placebo. Up to week 16, all patients received treatment as initially randomised. Based on the ACR 20% response (ACR20) at week 16, secukinumab doses were escalated at week 20 and patients were treated monthly until week 52. Responders continued on the same dose. Non-responders on 25 mg and 75 mg were titrated up to 150 mg. Those on 150 mg were titrated up to 300 mg. All patients on placebo were switched to secukinumab 150 mg, while all patients on secukinumab 300 mg stayed on the same dose. Details of the study design are presented in figure 1.

Study design.
Outcomes and assessments
The primary endpoint of this study was the ACR20 response rate at week 16 of each individual dose compared with placebo. Secondary efficacy endpoints included ACR20/50 responses at 2, 4 and 8 weeks, including ACR50/70 and disease activity score in 28 joints (DAS28) responses over time, treatment effect on ACR components including markers of inflammation (hsCRP and ESR) for 20 weeks.
Efficacy was assessed based on signs and symptoms according to ACR response criteria. DAS28 was derived from tender 28-joint and swollen 28-joint counts, ESR or hsCRP and patient's global assessment of disease activity at baseline and up to 16 weeks. Health-related quality of life was assessed using the health assessment quality–disability index (HAQ–DI).
Safety assessments included monitoring and recording all adverse events (AE) and serious adverse events (SAE), laboratory evaluations (haematology, blood chemistry, urinalysis and immunogenicity), vital signs, body weight, ECG and injection site reactions.
Regulatory and ethical review board approvals from competent authorities in each country were obtained for the study protocol. All patients signed an informed consent document, and the study was conducted in accordance with the Declaration of Helsinki and followed good clinical practice guidelines.
Statistical analysis
The full analysis set comprised all subjects to whom study drug was assigned and was the primary population for efficacy analyses. The safety set consisted of all subjects who received at least one dose of study treatment and was the primary population for safety analyses.
Demographic and baseline characteristics were summarised by treatment group, mean and SD were given for continuous variables, the SE of the mean was calculated when applicable and the categorical variables were summarised by absolute frequencies and percentages. The ACR20 responder rate at week 16 was compared against placebo for each of the secukinumab-treated groups based on a logistic regression model, with treatment, centre, baseline weight and baseline DAS28 as covariates. The estimate of OR and its 95% CI together with the corresponding p value was analysed for each treatment contrast. A generalised estimating equation method extending a logistic regression analysis was used to evaluate the treatment effect of secukinumab compared with placebo over time with treatment, centre, time point and subject by time point interaction as factors and baseline DAS28 and weight as covariates. Contrasts were estimated from the above model and odds ratios were determined with 95% CI. A last observation carried forward approach was used for imputing missing values. AE were summarised by absolute and relative frequencies, by treatment group.
The required sample size was determined assuming that the proportion of patients responding to placebo at week 16 would be 28%. A treatment difference in response rate of 35% was considered clinically relevant for active treatment versus placebo. With these assumptions, 37 randomly assigned patients were required in each group in order to have 80% power to show a significant difference between the secukinumab dose regimen and placebo at a 5% significance level in a two sided-test Fisher's exact test.
Results
Patients were screened (N=290) and randomly assigned (N=237) to one of the five treatment groups. Most patients (93.2%) completed the efficacy analysis of part I (week 16) of the study. A total of 16 patients discontinued the study before week 16 (figure 2).

Patient disposition.
Patient demographics and baseline characteristics
Patient demographics and baseline characteristics are presented in table 1. Most demographic attributes were balanced across treatment groups. Patients with previous exposure to biological agents were included in all groups (18.4–22.2%). Parameters of RA disease activity at baseline were overall well balanced. Approximately 67–80% of patients were rheumatoid factor positive and 72–88% were anticyclic citrullinated peptide antibody positive (table 1).
Patients’ baseline characteristics
Efficacy
Statistical significance for the primary efficacy endpoint, ACR20 response at week 16, was not achieved. ACR20 responses on secukinumab 25 mg, 75 mg, 150 mg and 300 mg dose groups compared with placebo were 34%, 46.9%, 46.5%, 53.7% versus 36.0%, respectively (table 2).
Number (%) of ACR20–ACR50–ACR70 responders by treatment and visit up to week 16, last observation carried forward (full analysis set)
Secondary outcomes
ACR50 response rates (at week 16) in the secukinumab 25 mg, 75 mg, 150 mg and 300 mg groups versus placebo were 15.1%, 18.4%, 20.9%, 17.1% versus 6% (table 2).
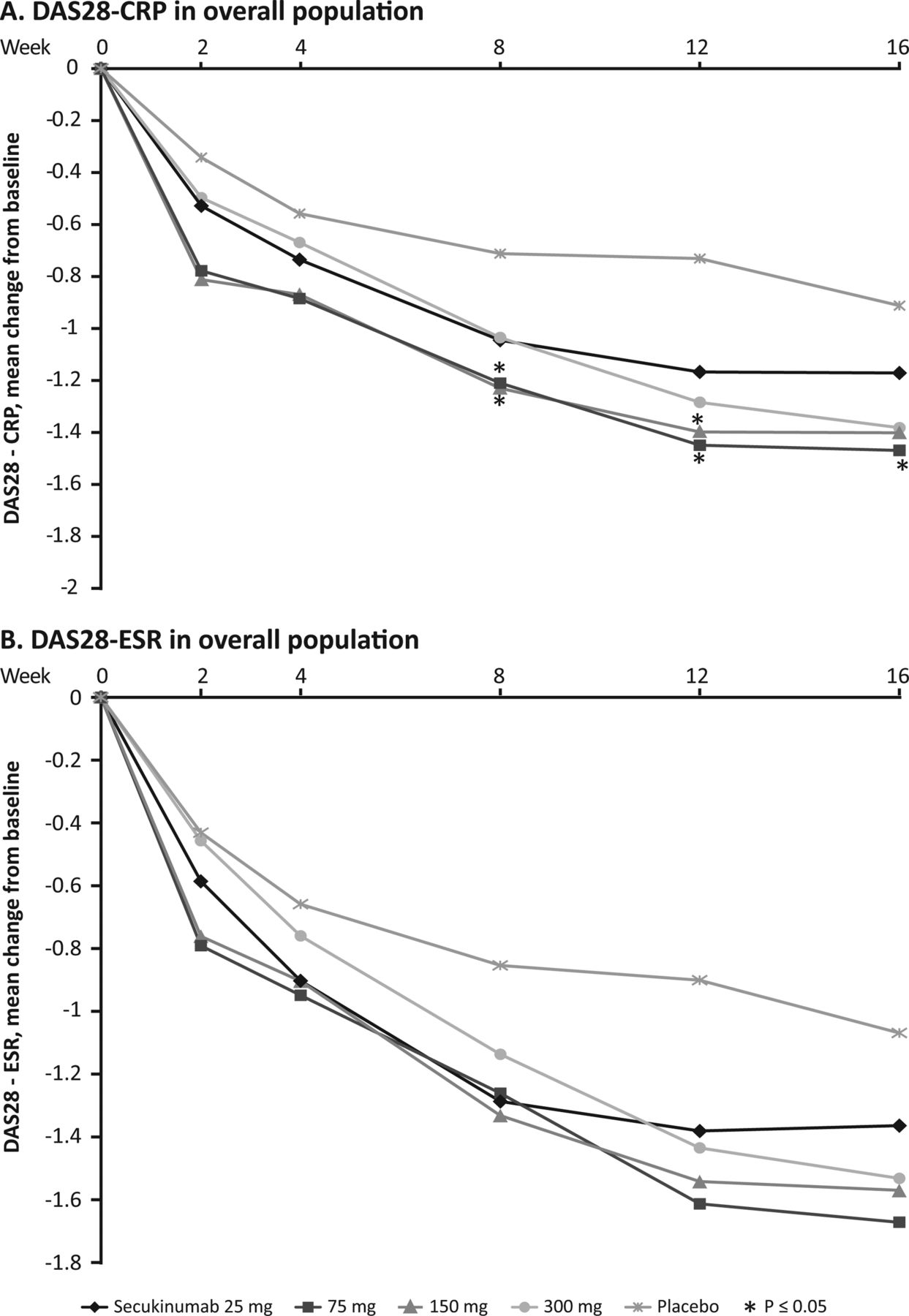
At week 16, a decrease in the DAS28–C-reactive protein (CRP) score from baseline was observed in all secukinumab dose groups versus the placebo group. This decline in DAS28–CRP score was observed as early as week 2 (figure 3A). At week 16, there was a meaningful reduction in the DAS28–CRP score of over 1.2 units with secukinumab 75–300 mg. A decrease was not evident with the 25 mg dose or with placebo. Based on repeated measures analysis of covariance, there was a significantly greater overall improvement from baseline in DAS28–CRP score over the 16 weeks with secukinumab 75 mg (difference in least squares (LS) mean (SE) −0.51 (0.243) (95% CI −0.99 to −0.03); p=0.0368) and 150 mg (difference in LS mean (SE) −0.48 (0.250) (95% CI −0.98 to −0.01); p=0.0543) doses compared with placebo. Similar to DAS28–CRP scores, a decrease in DAS28–ESR scores from baseline versus placebo was observed at week 2 and sustained up to week 16 (figure 3B). Serum hsCRP levels at week 16 were significantly reduced in the secukinumab 75 mg, 150 mg and 300 mg groups with differences versus placebo (difference in LS mean 95% CI) of −9.4 (−15.1 to −3.7) for 75 mg, −8.01 (−13.9 to −2.09) for 150 mg and −7.20 (−13.18 to −1.22) for 300 mg (p<0.05 for all). This was not the case for the 25 mg dose.

Change from baseline in disease activity score in 28 joints (DAS28)–C-reactive protein (A) and DAS28–erythrocyte sedimentation rate (B) over time in overall population groups (*=vs placebo p<0.05).
Reduction from baseline in the HAQ–DI score at week 16 was greater in all active dose groups compared with placebo but was not statistically significant with placebo-subtracted differences of −0.09 to −0.18 estimated by analysis of covariance. The mean reduction from baseline in tender joint count was higher with secukinumab 75 mg, 150 mg and 300 mg versus placebo (−5.8, −6.5, −6.4 vs −4.8, respectively) and the reduction in swollen joint count was higher with secukinumab 75, 150 and 300 mg versus placebo (−5.6, −5.3, −5.5 vs −4.1, respectively).
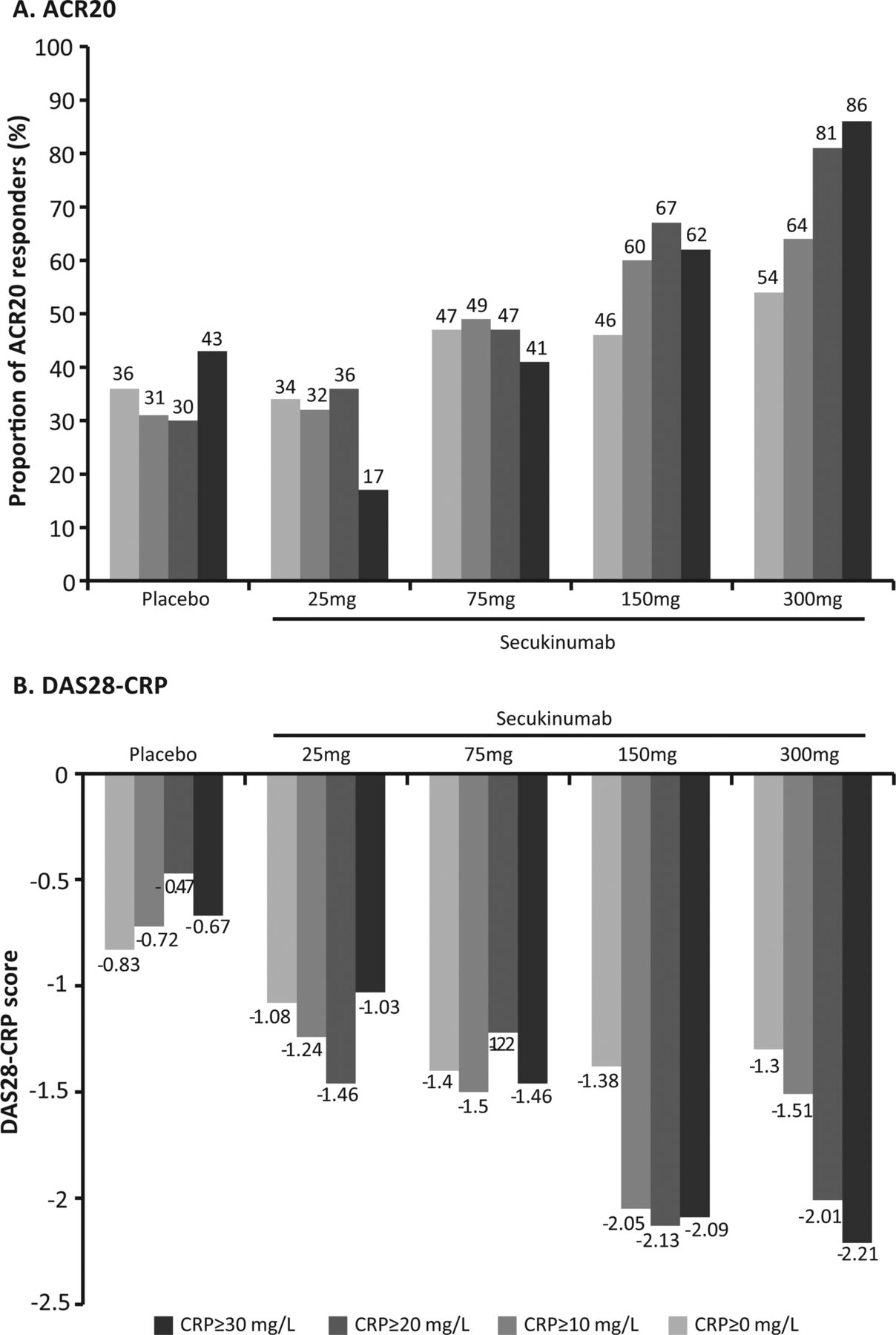
There was a dose-dependent relationship in ACR20 and DAS28–CRP responses at week 16 based on baseline hsCRP levels for secukinumab 150 mg and 300 mg compared with placebo (figure 4A,B). The 150 mg and 300 mg groups showed better efficacy with increasing levels of CRP (eg, ≥10 mg/l) for both endpoints; this trend was not observed for placebo, 25 mg and 75 mg secukinumab.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Dose–response relationship for American College of Rheumatology 20 (A) and disease activity score in 28 joints–C-reactive protein (CRP) (B) at week 16 by CRP levels at baseline.
Safety
The overall safety profile was comparable across the secukinumab and placebo groups. The rates of AE were similar across all treatment groups (table 3). Most of the AE were mild to moderate in severity and five patients discontinued the study due to AE before week 20, when discontinuations due to AE were comparable across groups: secukinumab 25 mg (n=3), 75 mg (n=0), 150 mg (n=0), 300 mg (n=1) and placebo (n=1) (figure 2). The rate of infections was higher in the secukinumab treatment groups compared with the placebo group (table 3); however, they were not dose-dependent. The infections reported across treatment groups consisted mostly of nasopharyngitis, upper respiratory tract infection, pharyngitis, sinusitis and urinary tract infection (table 3). A total of six SAE was reported up to week 20: secukinumab 75 mg (n=1; myocardial infarction); 300 mg (n=4; deep vein thrombosis, cholecystitis, pyelonephritis and pre-planned surgery for coxarthrosis) and placebo (n=1; vertebral fracture). One patient in each group except for 150 mg (n=2) reported injection site erythema, and four patients reported injection site pain (one case each in secukinumab 75 mg group and placebo and two cases in the 300 mg group). The laboratory assessments are detailed in table 3. Leucopenia was reported as AE in five patients (secukinumab 75 mg, n=4; 300 mg, n=1). Neutropenia was reported as an AE in one patient on secukinumab 75 mg and one in the placebo group. All cases of leucopenia and neutropenia were transient and there were no discontinuations due to leucopenia or neutropenia. There was one discontinuation due to anaemia in the placebo group. No immunogenicity could be confirmed in any of the patient samples. No deaths were reported up to week 20.
Safety data up to week 20
Discussion
This phase II, dose-finding study did not meet its primary objective. There was no statistically significant difference in ACR20 response at week 16 with secukinumab at any dose versus placebo. The data from this study do, however, highlight the difficulties of conducting phase II dose-ranging studies in RA in the current climate. Although not statistically significant at week 16, there is a trend suggesting that secukinumab at 75 mg, 150 mg and 300 mg subcutaneously monthly may provide some clinically meaningful benefit. Similarly differences between placebo and active drug appear more pronounced when looking at DAS28 scores over time than when looking at the primary endpoint in this trial. This has led the authors at least to question whether secukinumab might produce a clinically meaningful benefit in RA patients. The differences in DAS28 scores brings into question whether a different conclusion might be drawn if the DAS28 had been chosen as the primary endpoint rather than as the secondary endpoint.
At week 16, there was an improvement, but not statistically significant, in individual ACR components (including swollen and tender joint counts, patients’ and physicians’ global assessments of disease activity, hsCRP levels, ESR and HAQ–DI score) versus placebo, further supporting the potential of secukinumab 75 mg, 150 mg and 300 mg doses in the treatment of RA.
Overall, the interim analysis results failed to demonstrate benefit conclusively in the clinical efficacy of secukinumab. However, secondary endpoints suggest that secukinumab at doses of 75 mg, 150 mg and 300 mg subcutaneously every 4 weeks may be potentially therapeutic in the treatment of RA without the clear incremental benefit of the 300 mg dose. The 1 year data (up to week 52) analysis from this study and data from other studies of secukinumab in patients with RA should help to clarify these findings further along with evidence reported previously.13 ,22 ,23
Overall, the safety profile of secukinumab up to week 20 was consistent with that seen with other biological agents. There were no dose-dependent effects and the incidence of AE with secukinumab corroborates previous reports.19 AE causing drug discontinuation were infrequent and no deaths were reported up to week 20. As cytokine-targeted biological agents exert their effect by the inhibition of a specific pathway in the immune response, these agents can be associated with an increased incidence of infectious complications.24 In this study, the incidence of infections was slightly higher in some secukinumab dose groups versus placebo, but was not dose-dependent. The incidence of upper respiratory tract infections with secukinumab (2–4.7%) was comparable with placebo (4%) and other biological agents (2–14%).25 Overall, the safety data from this study, including AE, SAE, laboratory parameters and immunogenicity do not highlight any individual safety risk or particular pattern of event clustering. However, the number of patients in this study was small, making it difficult to interpret differences in the percentages of infrequent events, such as SAE. Large placebo controlled studies of secukinumab are ongoing and will serve to evaluate the safety of this compound better.
This phase II study has a number of limitations. The placebo-comparator duration of the study was short thus limiting comparison against placebo beyond 20 weeks. However, this design was implemented for ethical reasons to limit the duration for which patients were receiving methotrexate alone despite active disease. As a dose-ranging study with multiple arms, the total number of patients in any given arm was small. Had the sample sizes been larger, it might have been possible better to define clinically meaningful differences between active arms and placebo. As a corollary to this, the study was powered for a placebo rate of 28%; however, the rate seen was 36%, further limiting the power of the study. In general, the field has seen increased placebo responses in RA clinical trials in the biological era, particularly as studies have moved worldwide, and these may not have been adequately accounted for in the design of this study. The study enrolled a mixture of patients with and without previous exposure to biological agents; while this conceivably might have yielded some insight into the effect of secukinumab in each of the different populations, the study was not powered to assess subgroup effects. In addition, the importance of IL-17A relative to other cytokines in RA (eg, TNFα, IL-6, IL-1) as well as the relative importance of the Th17 versus Th1 pathways may differ between patients or disease stages. Accordingly, it will be crucial to identify biomarkers in patients with RA most likely to respond to IL-17A blockers and at which disease stage they are likely to derive the most benefit. Finally, safety data in this analysis are limited to week 20 thus not allowing for any conclusions on the longer-term safety of secukinumab.
Conclusions
In this study, the primary endpoint was not achieved. Treatment with secukinumab was well tolerated and did not generate a unique safety signature in this population over 20 weeks of exposure. The differences in response rates seen between active drug and placebo in ACR and DAS28 scores, particularly in 75 mg, 150 mg and 300 mg subcutaneously every 4-week arms, have led to the initiation of further studies to understand better the potential clinical utility of IL-17A blockade with secukinumab in RA patients.
Acknowledgments
The authors are grateful to the patients and study personnel who made this study possible and also wish to thank Youssef Saidi, Novartis Pharma AG, Basel, Switzerland, Kirtida Pandya Novartis Pharmaceutical Corporation, NJ, USA and Kalyan Pulipaka, Novartis Healthcare Pvt Ltd, Hyderabad, India for editorial assistance with this manuscript. The authors acknowledge Dr Karthinathan Thangavelu, Geneva, Switzerland for statistical contribution.
References
Footnotes
-
Contributors MCG, PD and TI substantially contributed to the study conception and design, acquisition of data, analysis and interpretation of data, drafting the article or revising it critically for important intellectual content and approved the final version of the article to be published. JAA, VM, HK, CEC and S-HL substantially contributed to the acquisition of data and approved all versions of the article to be published. SM, SH and HBR substantially contributed to the study conception and design, analysis and interpretation of data, drafting the article or revising it critically for important intellectual content and approved the final version of the article to be published. ED substantially contributed to the acquisition of data, drafting the article or revising it critically for important intellectual content and approved the final version of the article to be published. JS substantially contributed to the acquisition of data, analysis and interpretation of data and approved all versions of the article to be published.
-
Funding The study was financially supported by an unrestricted grant from Novartis Pharma AG, Basel, Switzerland.
-
Competing interests MCG has received grant research support and consulting fees from Novartis; JAA has received speaker fees, consulting fees and/or Honiara from Abbott, Amgen, Takeda, UCB and Wyeth/Pfizer; PD, JS, ED, VM, S-HL, CEC, HK and TI have nothing to disclose; HBR is an employee of Novartis; SH and SM are employees of Novartis and own stocks or stock options in Novartis.
-
Patient consent Obtained.
-
Ethics approval Regulatory and ethical review board approvals from competent authorities in each country were obtained for the study protocol.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
ClinicalTrials.gov identifier NCT00928512.