Article Text

Abstract
Objectives The objective of this study was to investigate the effect of the novel Janus kinase inhibitor CP-690,550 in fibroblast-like synoviocytes (FLSs) from patients with rheumatoid arthritis (RA).
Methods RA FLSs were isolated from tissue obtained by arthroplasty, cultured and serum-starved 48 h prior to stimulation. Messenger RNA and protein levels were determined by quantitative PCR and ELISA or multiplex bead assay, respectively. Phosphorylation of STAT (signal transducers and activators of transcription) proteins was determined by western blot.
Results Interleukin-6-induced phosphorylation of STAT1 and STAT3 was inhibited by CP-690,550 with IC50 values of 23 and 77 nM, respectively. Unexpectedly, although tumour necrosis factor (TNF) did not induce immediate phosphorylation of either STAT, CP-690,550 inhibited TNF-induced expression of several chemokines (IP-10, RANTES and MCP1) at the messenger RNA and protein levels. Chemokine expression was inhibited by cycloheximide, implying a need for de novo protein synthesis, and cycloheximide abolished the effect of CP-690,550 (tofacitinib). TNF induced early interferon (IFN) β expression and STAT1 phosphorylation beginning at 3 h, which was blocked by CP-690,550. The dependence of TNF-induced chemokine expression on type I IFN was confirmed in FLSs from mice lacking type I IFN receptors (IFNARs) and in RA FLSs using an IFNAR blocking antibody.
Conclusions The Janus kinase/STAT pathway in FLS is indirectly activated by TNF through autocrine expression of type I IFN, resulting in IFNAR engagement and production of T cell chemokines. These findings illuminate a novel role of CP-690,550 in the treatment of RA: the reduction of chemokine synthesis by FLS, thereby limiting recruitment of T cells and other infiltrating leucocytes.
Statistics from Altmetric.com
Introduction
CP-690,550, a selective inhibitor of the Janus kinase (JAK) family of receptor-associated tyrosine kinases,1 is currently in clinical development for the treatment of various autoimmune and inflammatory diseases including rheumatoid arthritis (RA). In rodent models of arthritis, CP-690,550 displayed considerable anti-inflammatory activity, as well as the ability to decrease bone and cartilage destruction.2 Clinical trials in RA demonstrated significant efficacy by CP-690,550 in multiple phase II trials3 4 and, as recently reported, in a phase III trial.5
The precise mechanism of action by CP-690,550 in RA is currently unclear. Several families of cytokines with potential relevance in RA signal through JAKs and might all be affected by CP-690,550, including the interferons (IFNs) and IFN-related cytokines, the common γ chain cytokines (interleukin (IL)-2, IL-7, IL-9, IL-15, IL-21, etc), as well as the gp130 family (including IL-6 and related proteins). JAKs associate with the cytosolic domain of cytokine receptors and, upon ligation of cytokine ligands, are brought into apposition leading to trans-phosphorylation and initiation of downstream signalling events, including the recruitment and phosphorylation of STATs (signal transducers and activators of transcription), which in turn drive gene transcription. Different cytokines signal through specific JAKs and STATs—for example, type I IFNs activate JAK1/TYK2, STAT1 and STAT2, whereas IL-6 activates JAK1, STAT1 and STAT3.6 In lymphocytes, stimulation with common γ chain cytokines leads to activation of JAK1/JAK3 and STAT5. Although CP-690,550 was initially thought to target JAK3 selectively,1 more recent data indicate the ability of the compound to inhibit JAK1, JAK2 and JAK3 and, to a lesser extent, the related TYK2 kinase in cell-free assays. In cellular assays, CP-690,550 preferentially inhibits cytokine signalling that requires JAK1 and/or JAK3 with lower potency against signalling dependent on JAK2 homodimers.7
Whereas the in vivo and ex vivo inhibition of T lymphocyte activity by CP-690,550 is well characterised,7,–,10 much less is known about the effect of CP-690,550 on other lineages involved in the pathogenesis of RA. One such cell of particular interest is the fibroblast-like synoviocyte (FLS), which plays a crucial role in joint destruction.11 The role of FLS as a target of CP-690,550 is consistent with its significant protection of bone and cartilage observed in rodent models of arthritis.2 Some cytokines that signal through JAK/STAT are known to activate FLS, including IL-6 and oncostatin M,12 13 and the receptor for IL-21 is present on FLS,14 although the effect of IL-21 itself on FLS remains uncharacterised. In addition, stimulation of Toll-like receptors on FLS activate an antiviral response that includes the synthesis of IFN and IFN-responsive genes.15 16 Therefore, the activation of JAK/STAT pathways in FLS and its inhibition by CP-690,550 were studied. Unexpectedly, we found that tumour necrosis factor (TNF) induces the expression of several lymphocyte-attracting chemokines by FLS through a CP-690,550-sensitive mechanism, which we identified as secondary autocrine stimulation by FLS-synthesised type I IFN.
Methods
Reagents
TNF, IFNβ and IL-1β were obtained from R&D Laboratories (Minneapolis, Minnesota, USA). IL-6, soluble IL-6 receptor (sIL6R) and IL-21 were from GenWay Biotech, Inc. (San Diego, California, USA). CP-690,550 (LC Laboratories, Woburn, Massachusetts, USA) was dissolved in dimethyl sulphoxide and kept at −20°C. A blocking mouse monoclonal antibody against the human type I IFN receptor chain 2 (clone MMHAR-2) was purchased from PBL InterferonSource (Piscataway, New Jersey, USA), and a corresponding isotype control was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, California, USA). All other reagents were from Sigma-Aldrich (St. Louis, Missouri, USA) unless otherwise noted.
Fibroblast-like synoviocytes
Human FLSs were cultured from the synovial tissues of patients with RA undergoing arthroplastic surgery, as previously described,17 after obtaining informed consent under approval from the University of California, San Diego Institutional Review Board. As in the study by Han et al,18 mouse FLSs were isolated from ankles and wrists of wild-type (wt) C57BL/6 animals as well as mice lacking the type I IFN receptor (Ifnar−/−),19 purchased from B&K Universal (East Yorkshire, UK) and back-crossed for 10 generations onto the C57BL/6 background. All FLSs were maintained in Dulbecco's Modified Eagle Medium supplemented with antibiotics, glutamine and 10% fetal bovine serum. Passages 4 through 8 were used in experiments. Cells were subjected to a 1–3-day reduced serum condition (0.1% fetal bovine serum) prior to stimulation to minimise baseline activity. CP-690,550 and cycloheximide were added to cultures at a final concentration of 0.1% dimethyl sulphoxide.
Western blot
FLS extracts were analysed by western blot as earlier described.20 Membranes were probed with antibodies against phospho-STAT1 (Tyr701) or phospho-STAT3 (Tyr705) or antibodies against corresponding total STATs, as well as with secondary antirabbit IgG-HRP (Cell Signaling Technology, Danvers, Massachusetts, USA). GAPDH was used as a gel-loading control (antibody from Santa Cruz Biotechnology, Inc.). Blots were developed with Immun-Star WesternC ECL substrate (Bio-Rad, Hercules, California, USA) and imaged on a VersaDoc imaging system (Bio-Rad), using QuantityOne software for image capture and densitometry.
Secreted protein assays
FLS supernatants were assayed by ELISA for MCP1 (eBioscience, San Diego, California, USA), MMP3 (GE Healthcare Biosciences, Piscataway, New Jersey, USA) and IP-10 (PeproTech, Rocky Hill, New Jersey, USA). Standard curves were constructed by regression line fitting on log(absorbance) versus log(concentration). Levels of cytokines and chemokines in supernatants were determined by Luminex multiplex analysis (Bio-Rad Bio-Plex) from four-parameter standard curve fits.
Gene expression assays
Total RNA isolated with RNA STAT-60 was reverse transcribed using random hexamer primers. The resulting complementary DNA was analysed by real-time TaqMan PCR using primer/probe sets from Applied Biosystems (Foster City, California, USA) using a cell-based standard approach as earlier described.21 Briefly, sample C(t) data were normalised to a standard curve generated from serial dilutions of complementary DNA derived from human FLS stimulated with IL-1β (human) or mouse FLS stimulated with polyinosinic:polycytidylic acid, generating cell equivalents. The ratio between biomarker cell equivalents and GAPDH cell equivalents was expressed as relative expression units. For IFNβ1, the ΔΔC(t) method was used due to lack of expression in the IL-1β-stimulated standard.
Statistical analysis
Data are reported as mean±SEM. RNA time-course data were analysed by two-way analysis of variance followed by contrast testing. All other experiments were analysed by one-way analysis of variance followed by Dunnett's post hoc test comparing control to all others or by paired Student t test, as appropriate. Real-time quantitative PCR and western blot data were log-transformed prior to analysis in order to approximate normality and variance equality. p<0.05 is considered significant.
Results
CP-690,550 inhibits IL-6-induced STAT1 and STAT3 phosphorylation in FLS
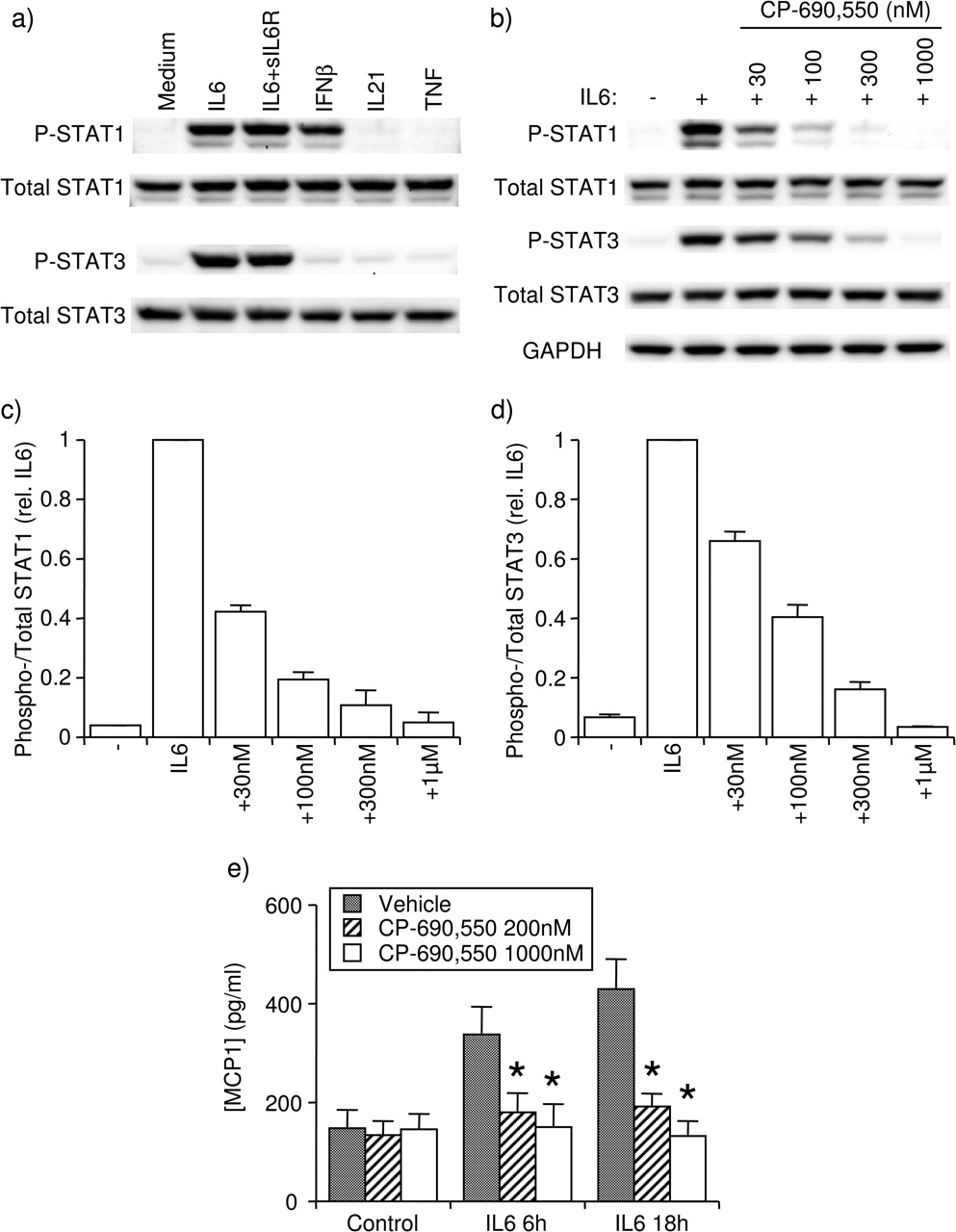
FLS from patients with RA expressed STAT1 and STAT3 protein (figure 1A,B). Treatment with IL-6 (20 ng/ml), in the presence or absence of sIL6R (100 ng/ml), induced tyrosine phosphorylation of STAT1 and STAT3 at 15 min, whereas IFNβ treatment phosphorylated only STAT1 (figure 1A). Neither IL-21 (50 ng/ml) nor TNF (10 ng/ml) had any effect (figure 1A). The JAK inhibitor CP-690,550 inhibited IL-6-induced phosphorylation of STAT1 and STAT3 in a dose-dependent fashion (figure 1B–D). On log–log graphs, IC50 for CP-690,550 inhibition of STAT1 and STAT3 phosphorylation was estimated to be 23 and 77 nM, respectively. Similar results were obtained with FLS from patients with osteoarthritis (OA, data not shown). In preliminary studies, treatment with IL-6+sIL6R rapidly induced MCP1 expression with peak messenger RNA levels at 1.5 h and returning to baseline by 5 h, whereas increased messenger RNA transcripts for other inflammatory mediators studied, including IL-8, MMP3, MCP1, IP-10, MIP1α and RANTES, were not detected following IL-6 treatment (data not shown). A corresponding increase in MCP1 protein secretion was observed at 6 and 18 h following addition of IL-6+sIL6R (figure 1E). CP-690,550 at 200 or 1000 nM completely prevented IL-6-induced MCP1 protein secretion (figure 1E). In further studies, CP-690,550 was used at 1000 nM unless otherwise indicated.

Effect of CP-690,550 on IL-6-induced responses in FLS. (A) Induction of phospho-STAT1 and phospho-STAT3 by various cytokines at 15 min. (B–D) Inhibition by CP-690,550 of IL-6-induced phosphorylation of STAT1 and STAT3, shown as (B) western blot representative of three experiments and overall average values for (C) STAT1 and (D) STAT3. (E) Inhibition of IL-6+sIL6R-induced MCP1 secretion by CP-690,550. Mean±SEM, n=3 RA FLS lines. *p<0.05 to veh by ANOVA/Dunnett's post hoc test. ANOVA, analysis of variance; FLS, fibroblast-like synoviocyte; IFN, interferon; IL, interleukin; RA, rheumatoid arthritis; sIL6R, soluble IL-6 receptor; STAT, signal transducers and activators of transcription; TNF, tumour necrosis factor; veh, vehicle.
CP-690,550 reduces TNF-induced synthesis of selected chemokines by FLS
The fact that TNF did not induce immediate phosphorylation of STAT1 or STAT3 (figure 1A) suggests that CP-690,550 should not affect TNF-induced gene expression. However, CP-690,550 significantly inhibited TNF-stimulated gene expression of selected chemokines, including MCP1, IP-10 and RANTES (figure 2A–F). Similar results were obtained with OA FLS (data not shown). The effect was observed at the RNA and protein levels and was especially prominent for IP-10 (figure 2C,D). Interestingly, inhibition of protein secretion by CP-690,550 was only observed at 18 h and not at 6 h, even though TNF had already begun to induce secretion of MCP1 and, to some degree, IP-10 at the earlier time point (figure 2B,D). The ability of CP-690,550 to inhibit gene expression was selective since TNF-induced IL-8 synthesis and secretion remained unchanged (figure 2G,H).

Inhibition of TNF-induced chemokine expression and secretion by CP-690,550 (1000 nM) in RA FLS. (A, C, E and G) Time course of TNF-induced mRNA expression in the presence and absence of CP-690,550. Mean±SEM, n=3 lines. *p<0.05 to TNF alone by two-way ANOVA and contrast testing on log-transformed data. (B, D, F and H) Chemokine concentration in cell culture media at 0, 6 and 18 h following TNF treatment in the presence and absence of CP-690,550. Mean±SEM, n=3–6 lines. *p<0.05 to veh by paired Student t test. ANOVA, analysis of variance; FLS, fibroblast-like synoviocyte; IL, interleukin; mRNA, messenger RNA; RA, rheumatoid arthritis; TNF, tumour necrosis factor; veh, vehicle.
In a separate experiment, the concentration of CP-690,550 required to inhibit TNF-induced IP-10 expression by FLS was determined. The IC50 for this effect was approximately 65 nM, similar to that earlier shown for IL-6-induced STAT phosphorylation (figure 1). These findings are consistent with CP-690,550 exerting its chemokine-inhibiting effect through blockade of a JAK/STAT pathway.
The ability of IL-1β, another inflammatory cytokine that activates FLS, to induce IP-10 was compared to that of TNF. As seen in supplementary figure 1, whereas IL-1β induced much lower expression of IP-10 than TNF, CP-690,550 still had a significant effect on this response.
CP-690,550 inhibits the action of a TNF-induced autocrine secondary mediator
The delayed effect on chemokine protein secretion by CP-690,550 suggested that TNF induces secretion of a secondary mediator, which then activates JAK/STAT-dependent secondary expression of IP-10, RANTES and MCP1. This hypothesis was tested in an experiment in which cycloheximide was used to block protein synthesis while leaving RNA expression intact. In support of the notion of an autocrine pathway, cycloheximide obliterated TNF-induced expression of IP-10 and RANTES (figure 3). In the case of IP-10 expression, the combination of CP-690,550 and cycloheximide had no further effect as compared with CP-690,550 alone (figure 3A), implying that the secondary mediator acts solely through JAK/STAT to induce IP-10. On the other hand, for RANTES, at 18 h, a minor part of the TNF-induced RANTES RNA synthesis was left intact by CP-690,550 but further inhibited by cycloheximide (figure 3B), suggesting that JAK/STAT-independent but protein-synthesis-dependent pathways are partly involved in RANTES expression.

TNF-induced chemokine expression is dependent on intact protein synthesis. CHX (10 µM) was used to inhibit de novo protein synthesis, and the expression of TNF-induced (A) IP-10 and (B) RANTES was studied in the presence and absence of CP-690,550 (1000 nM). Mean±SEM, n=3 RA FLS lines. *p<0.05 to veh by paired Student t test on log-transformed data. CHX, cycloheximide; FLS, fibroblast-like synoviocyte; RA, rheumatoid arthritis; TNF, tumour necrosis factor; veh, vehicle.
If TNF acts to induce a secondary mediator, which then in turn induces JAK/STAT pathways, delayed phosphorylation of STAT proteins should be observed following TNF treatment of FLS. This turned out to be the case since TNF stimulated significant phosphorylation of STAT1 (but not STAT3) at 3 h but not at earlier time points (figure 4A,B). As expected, CP-690,550 inhibited TNF-induced STAT1 phosphorylation at 3 h (figure 4C,D).

The secondary mediator induced by CP-690,550 might be IFNβ. (A and B) Late induction of phospho-STAT1, but not phospho-STAT3, in RA FLS by TNF. (C and D) Inhibition by CP-690,550 (CP, 1000 nM) of TNF-induced STAT1 phosphorylation. (E) Time course of TNF-induced IFNβ mRNA expression in the presence and absence of CP-690,550. (A and C) Western images representative of three experiments. (B, D and E) Mean±SEM of three RA FLS lines. *p<0.05 to (B) baseline by ANOVA/Dunnett's post hoc test and (D) veh by paired Student t test. ANOVA, analysis of variance; FLS, fibroblast-like synoviocyte; IFN, interferon; IL, interleukin; mRNA, messenger RNA; RA, rheumatoid arthritis; STAT, signal transducers and activators of transcription; TNF, tumour necrosis factor; veh, vehicle.
The fact that CP-690,550 had a particularly strong effect on IP-10 and the fact that STAT1 but not STAT3 phosphorylation was induced by TNF suggest an IFN-mediated effect. At 3 h through 24 h, TNF induced a fourfold to fivefold increase in IFNβ1 expression over baseline, which notably was not significantly inhibited by CP-690,550 (figure 4E). Together, these findings support the notion that TNF-induced type I IFN interacts with FLS receptors in an autocrine manner to activate JAK/STAT signalling pathways.
TNF induces type I IFN, which in turn induces IP-10 and RANTES
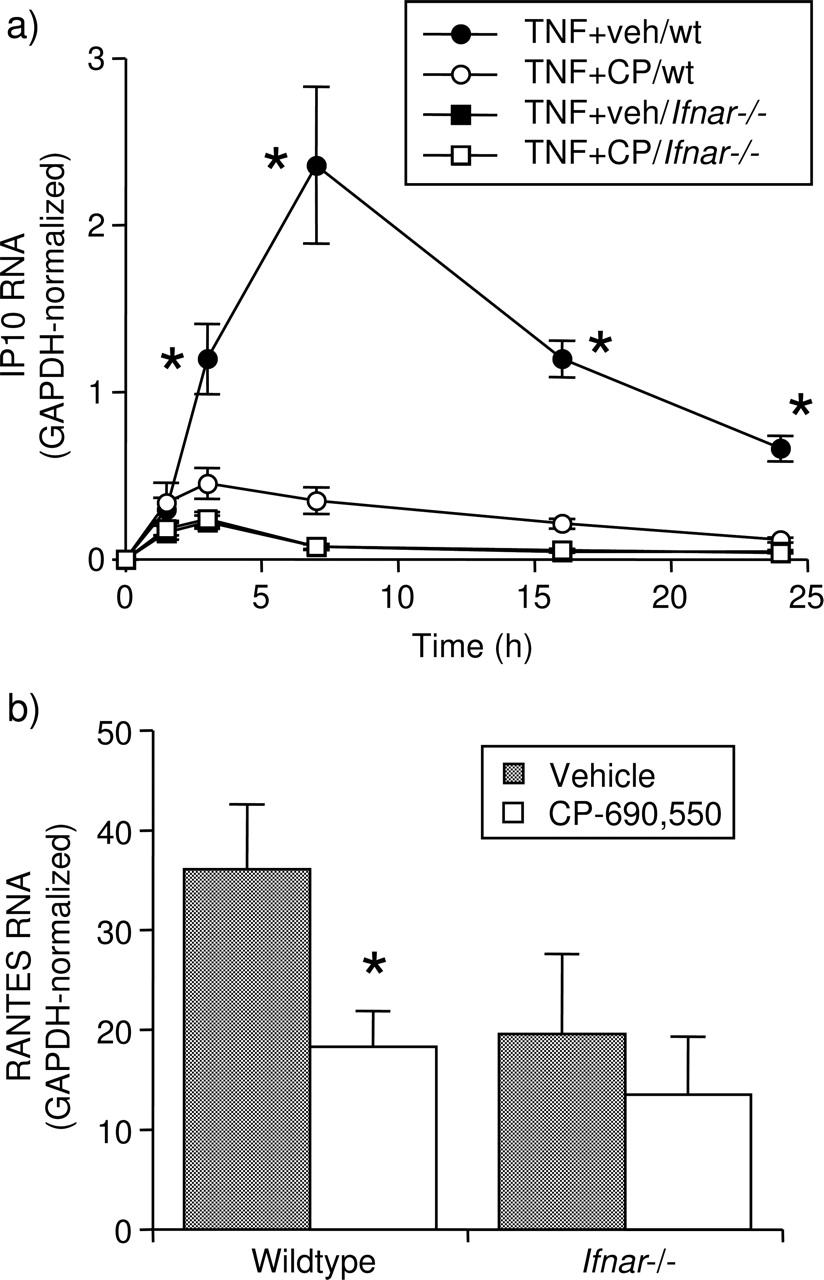
The role of type I IFN in the TNF-induced, JAK/STAT-mediated expression of IP-10 and RANTES was first determined in murine FLS lacking the type I IFN receptor (Ifnar−/−). The ability of CP-690,550 to inhibit TNF-induced IP-10 and RANTES expression was first tested in FLS from C57BL/6 wt mice to ensure that mouse FLSs act similarly to human RA FLSs. In both cases, substantial inhibition by CP-690,550 was observed (figure 5A,B). However, in FLS from Ifnar−/− mice, expression of IP-10 was significantly impaired compared to wt FLS, and the inhibitory effect of CP-690,550 was eliminated (figure 5A). The difference in RANTES expression between wt and Ifnar−/− FLS was less marked, but again, the effect of CP-690,550, evident in wt FLS, was lost in the FLS lacking the receptor (figure 5B). In parallel control experiments, TNF-induced Ifnb1 expression in Ifnar−/− FLS was not reduced compared to wt FLS (data not shown).

Type I IFN is required for the inhibition of TNF-induced chemokine expression by CP-690,550 in mouse FLS. (A) Time course of TNF-induced IP-10 mRNA expression after treatment with veh or CP-690,550 (CP, 1000 nM), in FLS from wt and type I IFN-receptor-deficient (Ifnar−/−) mice. Note that the results from Ifnar−/− FLS treated with veh or CP-690,550 overlapped, and only one of the two groups could be visualised. (B) Expression of RANTES mRNA at 24 h following treatment with TNF in the presence or absence of CP-690,550 in the same experiments as in (A). *p<0.05 to (A) TNF+CP-690,550 by two-way ANOVA and contrast testing and (B) veh by paired Student t test, on log-transformed data. ANOVA, analysis of variance; FLS, fibroblast-like synoviocyte; IFN, interferon; mRNA, messenger RNA; TNF, tumour necrosis factor; veh, vehicle; wt, wild type.
A blocking antibody to the type I IFN receptor was used to test the dependence of TNF-induced IP-10 expression on type I IFN in human FLS. When included in cultures along with TNF, it substantially diminished IP-10 gene expression (figure 6A) and inhibited IP-10 protein secretion by over 80% (figure 6B) as compared to isotype control. However, when CP-690,550 was included, the effect of the blocking antibody was no longer significant, suggesting that CP-690,550 and the antibody acted by blocking the same signalling pathway. Thus, taken together with earlier findings, these experiments demonstrated that TNF stimulates expression and secretion of type I IFN (eg, IFNβ) by FLS, which in turn interacts with receptors in an autocrine fashion to activate the JAK/STAT pathway and induce expression of IP-10 and, to a lesser degree, RANTES and MCP1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Type I IFN is responsible for the TNF-induced, CP-690,550-inhibited IP-10 expression in RA FLS. Cells were treated with veh or CP-690,550 (CP, 1000 nM) and with an antibody to type I IFN receptor (anti-IFNAR) or isotype control, and then stimulated with medium or TNF, and (A) IP-10 mRNA expression and (B) IP-10 concentration in cell culture media were studied at 18 h. Mean±SEM, n=3 RA FLS lines. *p<0.05 to veh by paired Student t test on (A) log-transformed or (B) untransformed data. FLS, fibroblast-like synoviocyte; IFN, interferon; mRNA, messenger RNA; RA, rheumatoid arthritis; TNF, tumour necrosis factor; veh, vehicle.
Discussion
The novel small-molecule pan-JAK inhibitor CP-690,550 is currently in late-stage clinical development for RA with promising results to date.3,–,5 In addition, it has shown promise as an immunosuppressive agent for use in transplantation.22 Due to its earlier perceived specificity for JAK3,1 which is mainly expressed in immune cells,23 CP-690,550 was chiefly thought of as targeting lymphoid and, to some degree, myeloid cells. More recently, it has become clear that CP-690,550 targets JAK1 and JAK2 with IC50 values in the same order of magnitude as that of JAK3.7 This has expanded the possible cellular targets of CP-690,550 in autoimmune and inflammatory disease. In the current report, we describe a novel mechanism of action for CP-690,550: interference with TNF-induced, IFNβ-mediated chemokine expression in FLS.
To begin with, the ability of CP-690,550 to interfere with JAK/STAT signalling in FLS was characterised using IL-6 as the stimulus. sIL6R was included to ensure maximum activation of IL-6-induced signalling pathways.24 25 The resulting IC50 values were similar to those earlier reported in human-cell-based assays conducted with T cells, where IL-6-induced STAT1 and STAT3 phosphorylation was inhibited by CP-690,550 at IC50 values of 54 and 367 nM, respectively,7 although in FLS, the difference in IC50 values for phosphorylation of STAT1 (23 nM) and STAT3 (77 nM) was less pronounced. Similar variations in cell-based IC50 values for CP-690,550 were earlier reported in T cells versus monocytes.7 Pharmacokinetic modelling in various human subject populations following twice daily dosing26,–,28 indicates that plasma concentrations are likely to exceed these IC50 values for the majority of a 24-h period (if not the entire period) depending on the dose, suggesting that the current results are relevant to the in vivo situation.
Although TNF did not immediately activate STAT1 or STAT3 in FLS, CP-690,550 significantly inhibited TNF-induced chemokine expression. The effect on IP-10 was particularly remarkable with inhibition in excess of 80–90%, with lesser although still significant inhibition of MCP1 and RANTES expression. Further experiments revealed this to be due to TNF-induced release of type I IFN, most likely IFNβ as earlier reported in FLS29 as well as in macrophages,30 although a role for IFNα cannot be excluded based on our current results. The extent of induction of IFNβ expression in FLS by TNF was only about fivefold over baseline expression, in agreement with earlier published results where the TLR3 ligand poly(I:C) had a much stronger effect on IFNβ expression than TNF.16 Nevertheless, TNF-induced type I IFN expression engaged FLS type I IFN receptors to induce IP-10 and other chemokines in an autocrine fashion.
IP-10, along with other chemokines, is thought to play a crucial role in the recruitment of T cells to the inflamed synovium. IP-10 levels are significantly elevated in RA synovial fluid and tissue, compared to OA, and over 90% of synovial T cells express the IP-10 receptor CXCR3.31 IP-10 synthesis in synovium is mainly localised to the lining and evident in macrophage-like synoviocytes and FLSs.32 Antibodies to IP-10 inhibit inflammation, T cell infiltration and bone destruction in experimental models of arthritis.33 34
The established pathological role of IP-10 in RA along with the fact that type I IFN, likely IFNβ, mediates TNF-induced IP-10 expression would seem to suggest that IFNβ plays a pathological role in the RA synovium. In fact, the effect of IFNβ in arthritis is controversial, and contradictory findings have been obtained in different species and models. In the mouse model of collagen arthritis, administration of IFNβ clearly confers protection,35 36 and IFNβ deficiency worsens disease.37 Similarly, in our hands, the K/BxN arthritis model has an accelerated course of disease in mice deficient in type I IFN receptors,19 whereas on the other hand, type I IFN is proinflammatory in other mouse arthritides such as viral double-stranded RNA-induced disease38 and a model of Lyme disease.39 In synovia from patients with RA, IFNβ is upregulated compared to OA and reactive arthritis and is particularly strongly expressed in FLS.40 The encouraging results using IFNβ treatment in mouse arthritis were initially duplicated in a small clinical trial in RA,41 but more rigorous randomised controlled trials did not reveal any activity of IFNβ in RA.42 43 In fact, reanalysis of the frequency of side effects reported in the van Holten et al43 study demonstrates that patients exposed to the higher dose of IFNβ experienced a significantly higher rate of aggravation of RA than the pooled placebo groups (19/68 vs 10/73, p<0.05), perhaps suggesting that IFNβ in fact may be proinflammatory in RA. IFNβ secreted by FLS and other cell types, as well as IFNα from synovial dendritic cells44 are likely to be involved in FLS chemokine synthesis in situ and to be susceptible to inhibition by CP-690,550.
In conclusion, our data demonstrate that CP-690,550 inhibits the expression of several lymphocyte-attracting chemokines, especially IP-10, by TNF-stimulated FLS. Further studies revealed that TNF induces IFNβ, which in turn acts in an autocrine fashion to stimulate JAK/STAT-dependent chemokine production. CP-690,550 inhibits type I IFN signalling in this system, thereby limiting chemokine induction. These findings should be placed alongside other potential actions by CP-690,550 in RA, including inhibition of IL-6-mediated pathways, as well as effects on T cell cytokine-induced signalling via JAK3. This novel mechanism of action by CP-690,550 in TNF-activated FLS is likely to be relevant to RA disease, especially since TNF is of large importance in RA pathology, as demonstrated by the clinical success of anti-TNF therapies in its treatment.
Acknowledgments
The authors are grateful to Josh Hillman and Lisa Ronacher for preparing the FLS.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
{kind=link}
Footnotes
-
Funding This work was supported, in part, by a research funding from Pfizer, Inc. (DLB), as well as by the University of California, San Diego Clinical and Translational Research Institute. MPC is funded through the Arthritis Foundation.
-
Competing interests SR and MPC have no competing interests to declare. GSF is a consultant to Pfizer, Inc. DLB received research funding from Pfizer, Inc.
-
Ethics approval This study was conducted with the approval of the University of California, San Diego Institutional Review Board.
-
Provenance and peer review Not commissioned; externally peer reviewed.