Article Text

Abstract
Objectives To evaluate the efficacy and safety of tabalumab, a human IgG4 monoclonal antibody that neutralises membrane and soluble B-cell activating factor (BAFF).
Methods This randomised, placebo-controlled study enrolled 1124 patients with moderate-to-severe systemic lupus erythematosus (SLE) (Safety of Estrogens in Lupus Erythematosus National Assessment- SLE Disease Activity Index ≥6 at baseline). Patients received standard of care plus subcutaneous study drug, starting with a loading dose (240 mg) at week 0 and followed by 120 mg every 2 weeks (120 Q2W), 120 mg every 4 weeks (120 Q4W) or placebo. Primary endpoint was proportion achieving SLE Responder Index 5 (SRI-5) improvement at week 52.
Results Clinical characteristics were balanced across groups. The primary endpoint was met with 120 Q2W (38.4% vs 27.7%, placebo; p=0.002), but not with the less frequent 120 Q4W regimen (34.8%, p=0.051). Although key secondary endpoints (time to severe flare, corticosteroid sparing and fatigue) were not met, patients treated with tabalumab had greater SRI-5 response rates in a serologically active subset and improvements in more stringent SRI cut-offs, SELENA-SLEDAI, Physician's Global Assessment, anti-double-stranded DNA antibodies, complement, total B cells and immunoglobulins. The incidences of deaths, serious adverse events (AEs), and treatment-emergent AEs were similar in the 120 Q2W, 120 Q4W and placebo groups, but depression and suicidal ideation, albeit rare events, were more commonly reported with tabalumab.
Conclusion SRI-5 was met with 120 Q2W and although key secondary endpoints were not met, numerous other secondary endpoints significantly improved in addition to pharmacodynamic evidence of BAFF pathway blockade. The safety profile for tabalumab was similar to placebo, except for depression and suicidality, which were uncommon.
Trial registration number NCT01205438.
- Systemic Lupus Erythematosus
- B cells
- Disease Activity
- Autoimmune Diseases
Statistics from Altmetric.com
Introduction
Systemic lupus erythematosus (SLE) is a heterogeneous autoimmune disease associated with chronic morbidity and premature death.1 Despite improvements in outcomes over the past 50 years, patients continue to experience chronic disabling symptoms, disease flares, complications associated with treatment (particularly with high-dose and long-term corticosteroid use) and gradual accumulation of organ damage.2 There are few drugs approved for treatment of SLE, and a substantial unmet medical need for evidence-based, effective and well-tolerated treatments remains.
B-cell activating factor (BAFF) is a tumour necrosis factor (TNF)-family ligand critical for B-cell development, differentiation and survival.3–5 Serum BAFF levels are frequently elevated in SLE and correlate with autoantibody production and disease activity.6 ,7 These observations prompted the development of belimumab, a BAFF inhibitor. Approved in a number of countries for the treatment of SLE, belimumab has been shown to reduce disease activity and autoantibody synthesis.8 ,9
Tabalumab, a fully human, subcutaneously administered, IgG4 monoclonal antibody, binds and neutralises both membrane and soluble BAFF.10 Two independent phase III studies, ILLUMINATE-1 and ILLUMINATE-2, evaluated the efficacy and safety of two doses of tabalumab plus standard of care (SoC) versus placebo plus SoC in patients with active SLE. We report the results of ILLUMINATE-2, where the primary efficacy measure was the SLE Responder Index 5 (SRI-5).9
Methods
Study design
ILLUMINATE-2 was a 52-week, multicentre, randomised, double-blind, placebo-controlled study. Patients were randomly assigned 1:1:1 (stratified by anti-double-stranded DNA (dsDNA) positivity and African descent) to receive subcutaneous tabalumab 120 mg every 2 weeks (120 Q2W) plus SoC, tabalumab 120 mg every 4 weeks (120 Q4W) plus SoC (administered by alternating 120 mg tabalumab injections and placebo Q2W), or placebo Q2W plus SoC. At week 0, the tabalumab groups received a 240 mg loading dose (2×120 mg injections), and the placebo group received two placebo injections. From week 0 through week 50, blinded treatments were administered Q2W. At week 52, patients either discontinued study drug and entered a safety follow-up period or began a multiyear, open-label, safety extension study of tabalumab. Data from the double-blind, 52-week period are included in this report.
Allowable SoC for SLE included non-steroidal anti-inflammatory drugs (unrestricted), corticosteroids, stable doses of antimalarials and/or stable doses of immunosuppressants; however, use of SoC for SLE was not required. Adjustments (increases or decreases) in corticosteroid dose were allowed during the first 24 weeks, after which the dose had to be less than or equal to the baseline dose and only reductions were allowed. No added or increases in antimalarials or immunosuppressant therapies were allowed at any time.
The protocol was approved by each institutional review board subject to applicable laws and regulations and ethical principles consistent with the Declaration of Helsinki. All patients provided written informed consent.
Entry criteria
Patients were eligible if they were 18 years or older, had a diagnosis of SLE (fulfilled 4/11 American College of Rheumatology criteria),11 had a positive antinuclear antibody (HEp-2 ANA; titre ≥1:80) test by the central laboratory during screening (patients with negative ANA, but positive anti-dsDNA, were retested before being excluded) and a Safety of Estrogens in Lupus Erythematosus National Assessment—SLE Disease Activity Index (SELENA-SLEDAI) score of ≥6.12 (See online supplementary material for detailed exclusion criteria.)
Endpoints
The primary endpoint was the proportion of patients achieving an SRI-5 response at week 52. This composite endpoint is defined as a ≥5 point improvement (reduction) in SELENA-SLEDAI score, no new British Isles Lupus Assessment Group 2004 (BILAG) index score of A or no more than one new BILAG B score and no worsening (increase of ≥0.3 points from baseline) in Physician's Global Assessment (PGA).9 Patients who did not meet all three clinical criteria, who added or increased antimalarial or immunosuppressant treatment, who could not adhere to corticosteroid-dosing requirements and/or who discontinued the study prior to week 52 were considered non-responders.
Key secondary endpoints included time-to-first severe SLE flare on the SELENA-SLEDAI Flare Index,13 the proportion of patients with reduction in corticosteroid dose by ≥25% to ≤7.5 mg/day prednisone (or equivalent) for ≥3 consecutive months from weeks 24 to 52 and change from baseline on the Brief Fatigue Index (BFI) at week 52.14 ,15
Other endpoints included change in proportion of SRI-5 responders over time; individual components of the SRI; the proportion of patients achieving SRI-4, and SRI-6 through SRI-10 at week 52, mean changes in SELENA-SLEDAI, PGA, SLEDAI-2K score, Systemic Lupus International Collaborating Clinics (SLICC) damage index, disease-related biomarkers (anti-dsDNA, C3 and C4), serum tabalumab concentrations, total B cells, B-cell subsets, BAFF levels, immunoglobulin concentrations, the Columbia-Suicide Severity Rating Scale (C-SSRS), the Quick Inventory of Depressive Symptomatology (QIDS-SR16) and other assessments of safety (serious adverse events (SAEs), treatment-emergent adverse events (TEAEs), adverse events (AEs) of special interest, vital signs, clinical laboratory data) at week 52. Additional pharmacodynamic and safety assessment details are provided in the online supplementary material.
Statistical analysis
All analyses of efficacy, serum tabalumab concentrations and biological activity were conducted on the intent-to-treat population. Safety analyses were applied to patients who received ≥1 dose of study drug. For the primary endpoint (SRI-5), a sample size of 380 patients per group was predicted to provide >95% power to detect a statistically significant difference of ≥14% between the tabalumab and placebo groups. Statistical methods used a two-sided significance level of 0.05 and included logistic regression for binary efficacy variables, Cox proportional hazard model for time-to-event variables, Fisher's exact for discrete variables and mixed model repeated measures (MMRM) or analysis of covariance for continuous variables. A graphical approach for multiple comparisons was used to control the overall type I error in the analysis of the primary and select secondary endpoints. Additional analyses were performed to identify baseline variables associated with response and to evaluate how response to tabalumab relative to placebo varied across different categories within a subgroup. Analyses of efficacy variables were adjusted for randomisation stratification factors (anti-dsDNA status and race) and region. For all non-SRI continuous measures, a last-observation-carried-forward (LOCF) approach was used.
Results
Patient disposition and baseline characteristics
Of the 2419 patients screened, 1124 (46%) were randomised and 872 (77.6%) patients completed the 52-week trial (figure 1). The percentage of patients who completed the trial and reasons for withdrawal were similar across groups. Baseline demographics, SLE disease duration and activity, concomitant medications and laboratory values were similar across treatment groups (table 1).
Baseline demographics and clinical characteristics

Flow diagram of patient disposition during the study. ITT, intent to treat; Q2W, every 2 weeks; Q4W, every 4 weeks.
Efficacy
The primary endpoint, proportion of patients meeting SRI-5 at week 52, was met by the 120 Q2W group (38.4% vs 27.7% in the placebo group, p=0.002), but not with the less frequent 120 Q4W regimen (34.8% vs 27.7%, p=0.051). For each component of the SRI, response rates were greater in the tabalumab 120 Q2W group versus placebo (table 2). Analysis of the time course of SRI-5 response revealed increasing differences between tabalumab 120 Q2W and placebo at week 24 (figure 2A), the time point at which corticosteroid dose had to be equal to or lower than baseline with no subsequent increases. These differences were statistically significant at weeks 16, 40, 48 and 52.
Clinical and biomarker outcomes
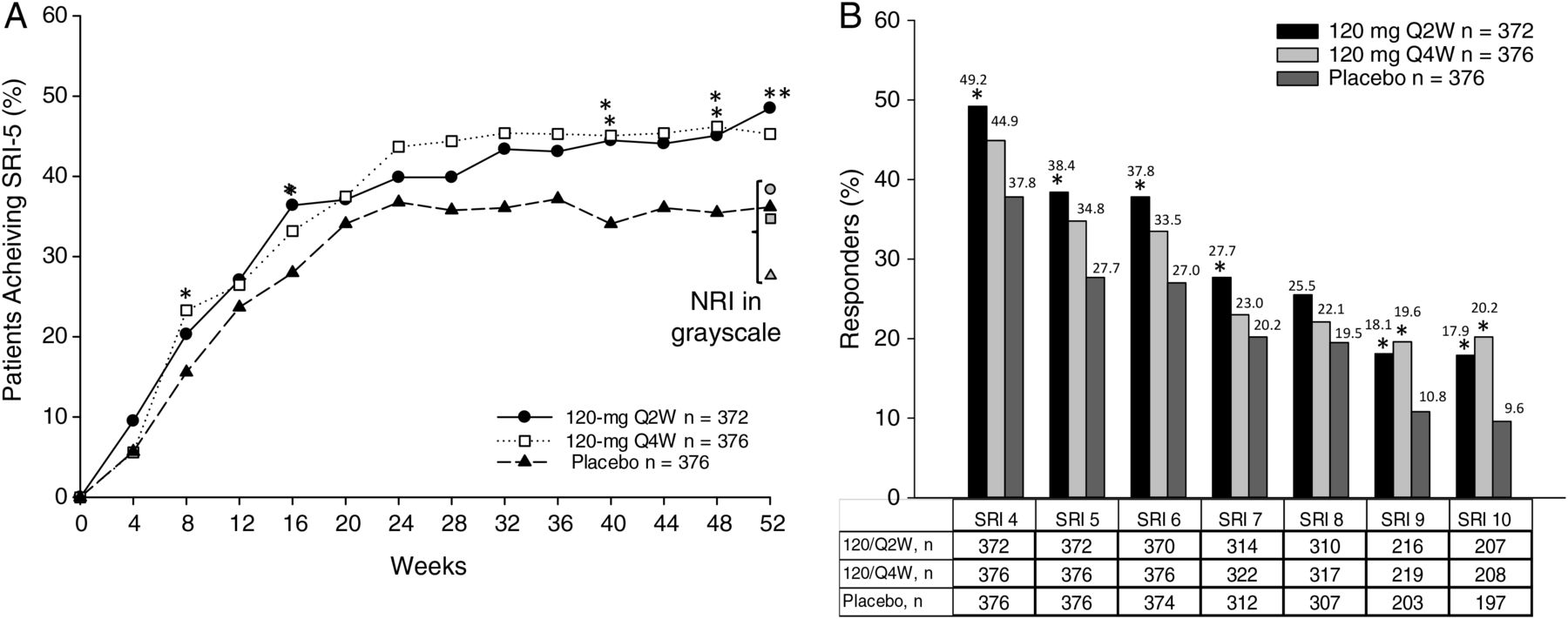
SRI-5 observed response rates over the 52-week treatment period and SRI-5 (NRI) primary endpoint at Week 52. The primary endpoint analysis included patients who achieved SRI-5 endpoint without violating concomitant medication rules. Patients who did not meet all three clinical criteria of the SRI, who added or increased antimalarial or immunosuppressant treatment, who could not meet the steroid-dosing requirements and/or who discontinued the study prior to week 52, were considered non-responders. For observed response rates, missing data were not imputed as non-response. For the pre-specified primary endpoint analysis, NRI was used to impute missing response data as non-response for the primary endpoint of SRI-5 at Week 52. p Value based on OR was estimated from logistic regression model. (B) SRI-4 through SRI-10 response rates at week 52. SRI-6–SRI-10 was performed in subset of patients who entered study with ≥SLEDAI6–SLEDAI10. NRI was used to impute missing response data for the endpoints of SRI-4 to SRI-10. *p≤0.05 versus placebo; **p≤0.001 versus placebo. Symbols in grayscale represent 52-week endpoint data calculated using a NRI, circle=120 Q2W, square=120 Q4W and triangle=placebo. NRI, non-responder imputation; Q2W, every 2 weeks; Q4W, every 4 weeks; SLE, systemic lupus erythematosus; SLEDAI, SLE Disease Activity Index; SRI, SLE Responder Index; SRI-4 through SRI-10, SLE Responder Index 4 through 10.
None of the key secondary endpoints (ie, time-to-first severe SLE flare, corticosteroid sparing effects and the change from baseline in BFI) were significantly different when comparing tabalumab and placebo (table 2).
In analyses at week 52 using different SRI cut-offs, the 120 Q2W group had a significantly higher percentage of responders than placebo on most measures (ie, SRI-4, -6, -7, -9 and -10), whereas the 120 Q4W group met the most stringent SRI-9 and SRI-10 responses relative to placebo (figure 2B). At week 52, reduction in SELENA-SLEDAI was greater in the 120 Q2W group (least squares (LS) mean: −5.5) than in the placebo group (LS mean: −4.5, p=0.002). Similar results were achieved in the 120 Q4W group (LS mean: −5.1) versus the placebo group (LS mean: −4.5, p=0.039).
The effect of tabalumab was also evaluated in patients who were anti-dsDNA-positive and who had low C3 and C4 levels at baseline with a significantly higher rate of SRI-5 response in the 120 Q2W group (41.2% vs 25.6% in the placebo group, p=0.015) and the 120 Q4W group (38.0%, p=0.036).
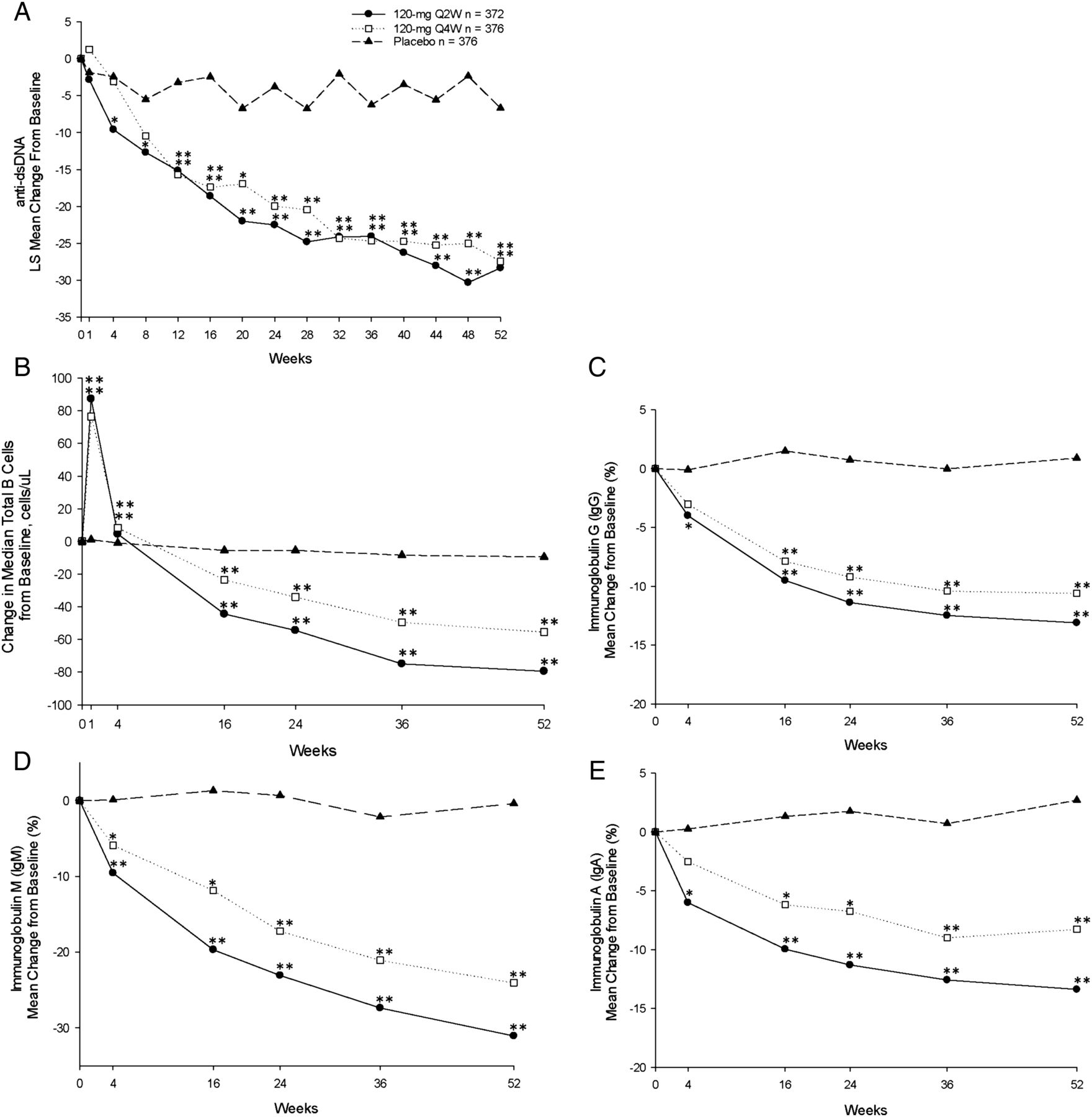
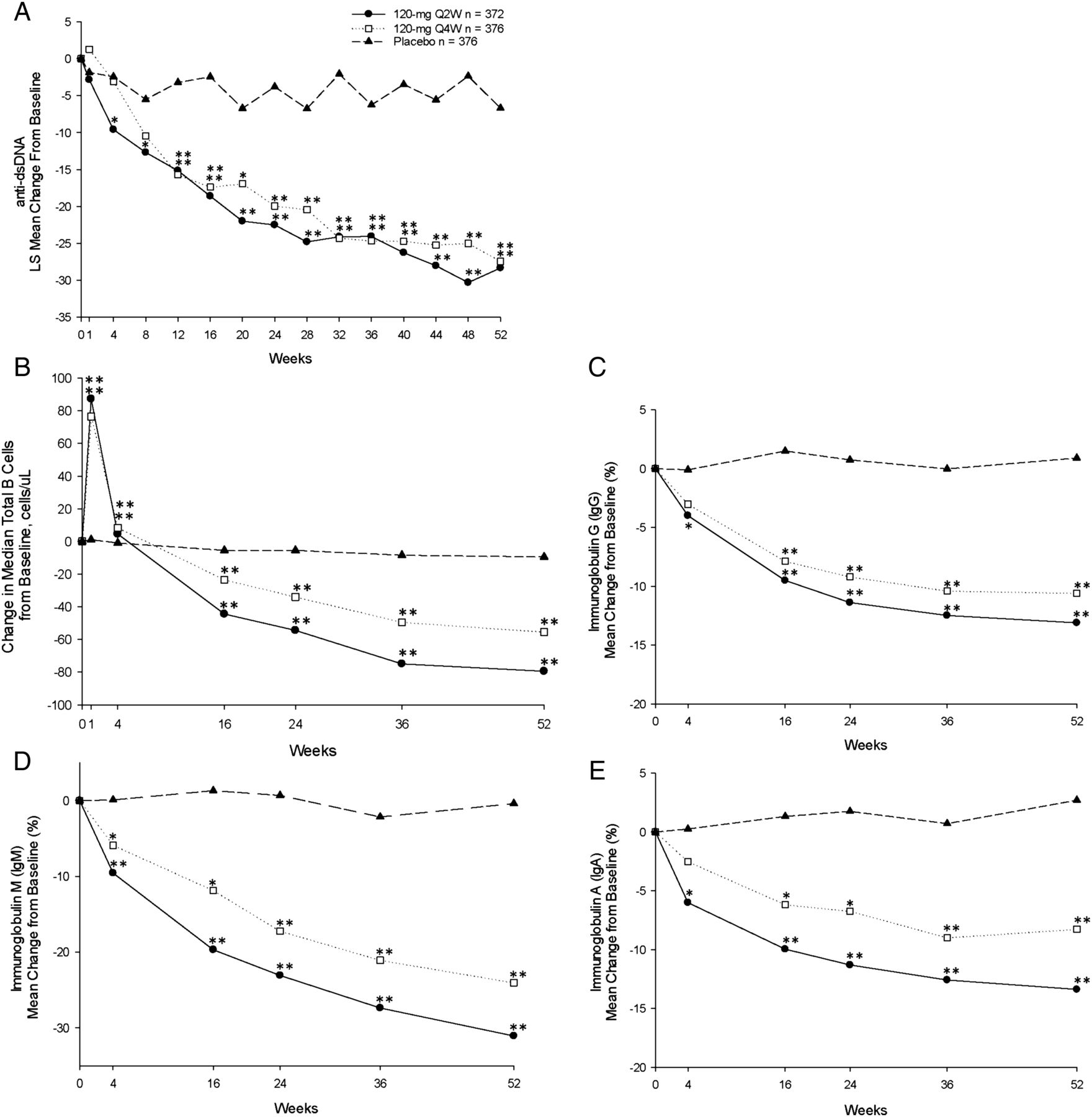
Anti-dsDNA levels decreased in both tabalumab groups as early as week 4 and continued to decrease, remaining well below baseline levels through week 52 (figure 3A) and were significantly different from placebo from week 12 to week 52 (table 2). Of the patients with anti-dsDNA of ≥30 IU/mL (positive) at baseline, 12% in both the 120 Q2W and 120 Q4W groups versus 7% in the placebo group were negative (<30 IU/mL) at week 52.
{kind=link}
{kind=link}
{kind=link}
Measures of biological activity. (A) Mean change in anti-dsDNA over the 52-week treatment period. (B) Mean change in total B cells over the 52-week treatment period. (C–E) Mean change in serum IgG, IgM and IgA, respectively, over the 52-week treatment period. *p≤0.05 versus placebo; **p≤0.001 versus placebo. dsDNA, double-stranded DNA; Q2W, every 2 weeks; Q4W, every 4 weeks.
In the Q2W dose group, increases in C3 and C4 were observed that were significantly greater than placebo at week 52; in the 120 Q4W group, the increase in C4 was significant, but the increase in C3 only neared significance at week 52 (table 2). Of the patients with low C3 and/or C4 at baseline, post-hoc analysis showed that 37% in the 120 Q2W group versus 22% in the placebo group (p=0.001) and 31% in the 120 Q4W group versus 22% in the placebo (p=0.046) group normalised at week 52.
In prespecified subgroup analyses, the following patient attributes did not have a statistically significant impact on response: sex, age, region, anti-dsDNA status, complement, race, ethnicity, disease severity, PGA score, SLICC score, severe seropositivity, concomitant medication use, number of prior treatment failures, comorbidities, time since onset of SLE or BAFF concentration.
Pharmacodynamics
Median total B-cell counts initially increased at week 1 (120 Q2W: 87.3 cells/µL; 120 Q4W: 76.5 cells/µL; placebo: 1.0 cells/µL; p<0.001, each comparison) and then decreased below baseline values through week 52 (LOCF) (120 Q2W: −76.3 cells/µL; 120 Q4W: −50.0 cells/µL; placebo: −8.0 cells/µL; p<0.001, each comparison) (figure 3B). Statistically significant decreases in serum immunoglobulins were observed at each time point over the 52-week treatment period (figure 3C–E). The number of patients who dropped below the normal range in the 120 Q2W, 120 Q4W and placebo groups was as follows: IgA: 3 (0.8%), 6 (1.7%), 1 (0.3%); IgG: 4 (1.1%), 3 (0.8%), 3 (0.8%); IgM: 61 (18.4%), 48 (14.8%), 13 (3.8%), respectively.
Pharmacokinetics
Tabalumab concentrations shifted higher in the 120 Q2W group compared with the 120 Q4W group; however, there was overlap between the two dosing regimens (data not shown).
Safety and tolerability
There were no differences in the number of deaths, the frequency of SAEs or TEAEs or the number of patients who discontinued due to an AE between groups (table 3). The majority of TEAEs were mild to moderate in severity.
Safety and tolerability
Nine pregnancies were reported (table 3). Of the six pregnancies reported with live births, there were no fetal abnormalities or malformations noted (120 Q2W, n=2; 120 Q4W, n=1; placebo, n=3) and no maternal complications reported. One terminated pregnancy (120 Q2W) and one spontaneous abortion (120 Q4W) were reported. Outcome of pregnancy was not reported for one patient in the placebo group.
The incidences of serious infections and severe infections were similar in the tabalumab and placebo groups (table 3). Opportunistic infections of interest included herpes zoster, which was reported in 6 patients (1.6%) in the 120 Q2W group, 10 patients (2.7%) in the Q4W group and 6 patients (1.6%) in the placebo group, and pulmonary tuberculosis, which was reported in 1 patient (0.3%) in the placebo group. Oral candidiasis was reported in four patients (1.1%) in the 120 Q2W group, five patients (1.3%) in the Q4W group and seven patients (1.9%) in the placebo group; however, no unexpected fungal opportunistic infections were reported. More patients in the 120 Q2W group reported injection-site reactions (9.4%) than in the 120 Q4W (7.5%) and placebo groups (5.1%). No anaphylactic events were reported; hypersensitivity reactions occurred in 9 patients in the 120 Q2W group, 11 patients in the 120 Q4W group and 4 patients in the placebo group, the majority of which were mild or moderate in severity. The incidence of adjudicated major adverse cardiovascular events was similar in the tabalumab and placebo groups, and malignancy was reported in four patients in the 120 Q4W group and two patients in the placebo group (table 3).
More patients receiving tabalumab than placebo reported depression (120 Q2W: 18 (4.9%; p=0.013); 120 Q4W: 21 (5.6%; p=0.003); placebo: 6 (1.6%)) (table 3). The number and percentage of treatment-emergent suicidal ideation on the C-SSRS was also higher with tabalumab (120 Q2W: 7 (3.0%; p=0.037); 120 Q4W: 12 (5.3%; p=0.001); placebo: 1 (0.4%)). All but two patients recovered (no report of suicidal ideation at subsequent visit): one patient (placebo) discontinued due to an SAE of butterfly rash and reported an adverse event of suicidal ideation that resolved, and one patient (Q4W) discontinued due to lack of efficacy and no additional follow-up data are available. No important differences in laboratory abnormalities across treatment groups were noted. The frequency of treatment-emergent antidrug antibodies was low, and no neutralising antidrug antibodies were identified (table 3).
Discussion
In this randomised, placebo-controlled trial of 1124 patients with active SLE, those receiving 120 Q2W tabalumab met the primary endpoint, SRI-5 (38.4% vs 27.7% (placebo), p=0.002). A dose–response was suggested by fewer patients meeting this endpoint when treated Q4W (34.8%, p=0.051 vs placebo). At baseline, tabalumab or placebo was added to a variety of SoC therapies, the impact of which on BAFF signals is unknown. Furthermore, major adjustments (including increases) in corticosteroid dose were allowed in the first 24 weeks, which may have affected BAFF levels.6 Similar to other trials allowing significant background medications, a high placebo response rate was expected. Whether forced steroid tapering or withdrawal of background treatments would increase tabalumab treatment effect remains unknown.8 ,9
Analyses of three secondary endpoints were multiplicity controlled: time-to-first severe SLE flare, corticosteroid sparing and the change from baseline in fatigue. Tabalumab did not meet these three endpoints, although active treatment had better outcomes than placebo on some measures. For example, the corticosteroid sparing endpoint was achieved by 13.9% in placebo group versus 22.5% in 120 Q2W (p=0.051) and 17.5% in 120 Q4W (p=0.342) groups. Steroid tapering was not mandated in improved patients. Since no other rescue was allowed after 6 months, incentive to reduce steroids was likely decreased, limiting insight into the potential of tabalumab to spare corticosteroids. Other secondary endpoints did show statistically significant differences between groups. More patients in the tabalumab group had improved SLEDAI-2K, SELENA-SLEDAI and PGA scores compared to placebo (table 2).
Tabalumab had significant impact on its intended immunological target with decreases in mean total B cells, immunoglobulins and anti-dsDNA antibodies. However, given the heterogeneity of SLE, the optimal dose remains unknown either for the cross-section of international patients who were studied or for individual patients. Clinical observations from this trial and from the ILLUMINATE-1 study (Ann Rheum Dis, submitted March 2015) suggest that different dosing strategies might be helpful for different patients, underscoring the importance of further responder analyses and pharmacodynamic assessments of BAFF inhibitors.
When the analysis was restricted to patients with baseline anti-dsDNA antibodies and low complement, the response rates increased slightly in patients treated with tabalumab and fell with placebo, increasing the treatment effect size. Anti-dsDNA antibodies and low complement are associated with higher BAFF signalling and could help define those more likely to respond to treatment with BAFF inhibitors; this is also a known subpopulation with more severe disease.16–19 In this study, BAFF concentrations were increased by tabalumab, but the assay could not distinguish between unbound (presumably bioactive) BAFF molecules and complexes between BAFF and tabalumab.
The SRI-5 endpoint creates a high bar for efficacy by incorporating a stringent definition for improvement (SLEDAI 5-point drop) with sensitive endpoints to ensure lack of worsening in any organ (BILAG and PGA). The SLEDAI alone slightly increases discrimination between treatment and placebo (table 2) but is a less-rigorous determinant of efficacy. Figure 2 compares response rates using the SRI-4 (a less-stringent definition of response than the primary endpoint) and the SRI-5 through SRI-10 (serially raising the bar for response). These restrictive endpoints produced lower response rates overall, but differences between tabalumab and placebo increased in those patients with the highest baseline disease who qualified for the SRI-9 and SRI-10 analyses. Therefore, despite aggressive immune suppression and rescue treatments in this trial, placebo response rates were not as problematic when limiting analysis to the most severe patients. Similar observations were also made in post-hoc analyses of belimumab data.9 ,20
Another phase III study of tabalumab, ILLUMINATE-1 (Ann Rheum Dis, submitted March 2015), did not meet the SRI-5 primary endpoint. There are no obvious differences between the populations in these trials; they were both worldwide trials with similar baseline medications and subject demographics. However, ILLUMINATE-1 stipulated that new, increased or decreased SoC medications would define a patient as non-responsive, whereas only new or increased medications in the current trial determined non-response. A major difference between the ILLUMINATE and phase III belimumab studies is that the two belimumab trials were conducted in entirely separated areas of the world.9 ,8 When the ILLUMINATE-2 definition of response is used, and when the SRI-4 outcome used in the belimumab trials is examined (see figure 2), differences between the ILLUMINATE and belimumab outcomes are diminished.
The overall safety of tabalumab was similar to the placebo group. Although depression and suicidal ideation were rare, they were more common in patients treated with tabalumab. There was also a numerical increase in depression in one phase III belimumab study.9 BAFF is expressed in the central nervous system (CNS) of patients with multiple sclerosis (MS) and neuromyelitis optica (an MS spectrum disease with strong immunological overlap with SLE),21 ,22 has been reported in the CNS of patients with SLE and is more enhanced in those with neuropsychiatric involvement.23 Anxious behaviour has also been reported in an autoimmune BAFF transgenic murine model.24 Since BAFF supports in vitro neural cell survival, a drop in BAFF signalling could alter neural function and change mental state.25 Although there does not appear to be a high risk of suicide in patients with SLE taking BAFF inhibitors, it seems reasonable to follow patients closely for symptoms of depression.
This double-blind, placebo-controlled trial of tabalumab met its primary endpoint while demonstrating pharmacodynamic evidence of BAFF pathway blockade. Although multiplicity-controlled secondary endpoints were not met, a number of other clinical and immunological endpoints provide justification for further study of this pathway.
Acknowledgments
The authors would like to thank all of the ILLUMINATE-2 investigators for their participation; Rebecca Taha, Steven Watts, Tammy Forrester and Allen Nyhuis of Eli Lilly and Co. (Indianapolis, IN) for assisting in statistical analyses and Kelly Guerrettaz and Cindi Wood of inVentiv Health Clinical (Princeton, NJ) for assisting in manuscript writing and editing, respectively.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Lay summary
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Tore K Kvien
Correction notice This article has been corrected since it was published Online First. Figure 2 has been corrected.
Contributors JTM has participated in study conception and design, acquisition of data, analysis and interpretation of data and drafting and critical revision of the manuscript. RFvV, WS, CL, P-YB and CD have participated in study conception and design, analysis and interpretation of data and drafting and/or critical revision of the manuscript. MM-C has participated in study conception and design, statistical analysis and interpretation of data and critical revision of the manuscript. JPB, RAF, TD and PWA have participated in the analysis and interpretation of data and drafting and/or critical revision of the manuscript.
Funding Funding for this study and this publication: Eli Lilly and Company.
Competing interests JTM has been a speaker for GlaxoSmithKline, has received a research grant from GlaxoSmithKline and has been a consultant for Eli Lilly and Company, GlaxoSmithKline, Anthera Pharmaceuticals and EMD Serono. RFvV has been a consultant for AbbVie, Biotest Pharmaceutical Corporation, Bristol-Meyers Squibb, Crescendo Pharmaceutical Company, GlaxoSmithKline, Janssen Pharmaceuticals, Eli Lilly and Company, Merck & Co., Pfizer, Roche, UCB and Vertex and has received research grants from AbbVie, Bristol-Meyers Squibb, GlaxoSmithKline, Pfizer, Roche and UCB. JPB has been a consultant for Eli Lilly and Company and GlaxoSmithKline and has received a research grant from GlaxoSmithKline. RAF has been a consultant for Eli Lilly and Company. WS has been a consultant for Celgene, Eli Lilly and Company. and Janssen (less than US$10 000 for each). MM-C, CD, PWA, CL and P-YB are employees and stockholders of Eli Lilly and Company. TD has received honoraria for scientific advice (less than US$10 000) from Eli Lilly and Company and has received study support and honoraria for scientific advice by UCB, Roche/Chugai, Sanofi.
Ethics approval Institutional review board at each study site.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Editorial
- Clinical and epidemiological research
- Electronic pages