Article Text

Abstract
Background Psoriatic arthritis (PsA) is a chronic inflammatory disease, characterised by synovitis and destruction of articular cartilage/bone. Janus-kinase and signal transducer and activator of transcription (JAK-STAT) signalling pathway is implicated in the pathogenesis of PsA.
Objectives To examine the effect of tofacitinib (JAK inhibitor) on proinflammatory mechanisms in PsA.
Methods Primary PsA synovial fibroblasts (PsAFLS) and ex vivo PsA synovial explants were cultured with tofacitinib (1 µM). PhosphoSTAT3 (pSTAT3), phosphoSTAT1 (pSTAT1), suppressor of cytokine signaling-3 (SOCS3), protein inhibitor of activated Stat3 (PIAS3) and nuclear factor kappa B cells (NFκBp65) were quantified by western blot. The effect of tofacitinib on PsAFLS migration, invasion, Matrigel network formation and matrix metallopeptidase (MMP)2/9 was quantified by invasion/migration assays and zymography. Interleukin (IL)-6, IL-8, IFN-gamma-inducible protein 10 (IP-10) monocyte chemoattractant protein (MCP)-1, IL-17, IL-10, MMP3 and tissue inhibitor of metalloproteinases 3 (TIMP3) were assessed by ELISA.
Results Tofacitinib significantly decreased pSTAT3, pSTAT1, NFκBp65 and induced SOCS3 and PIAS3 expression in PsAFLS and synovial explant cultures (p<0.05). Functionally, PsAFLS invasion, network formation and migration were inhibited by tofacitinib (all p<0.05). In PsA explant, tofacitinib significantly decreased spontaneous secretion of IL-6, IL-8, MCP-1, MMP9/MMP2, MMP3 (all p<0.05) and decreased the MMP3/TIMP3 ratio (p<0.05), with no effect observed for IP-10 or IL-10.
Conclusions This study further supports JAK-STAT inhibition as a therapeutic target for the treatment of PsA.
- Psoriatic Arthritis
- Synovitis
- Inflammation
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Psoriatic arthritis (PsA) is a chronic inflammatory arthritis associated with psoriasis (Ps) and characterised by synovitis and progressive destruction of articular cartilage and bone. One of the earliest events is new vessel formation and invasion resulting in a self-perpetuating and persistent infiltration of leucocytes, transforming the synovium into an aggressive tumour-like ‘pannus'. Previous studies have demonstrated distinct macroscopic vascularity in the PsA joint characterised by elongated, tortuous vessels which is associated with increased expression of cytokines, angiogenic growth factors and decreased cell apoptosis.1 ,2 This facilitates PsA synovial fibroblasts (FLS) to invade adjacent cartilage and bone resulting in joint destruction.
PsA synovium is the target of interplay for many proinflammatory cytokines and growth factors, with key circulating mediators controlling cell traffic from the blood stream into the underlying tissue. Recently developed agents for PsA target IL12p40, interleukin (IL)-6 and IL-17,3–5 several of which signal through the Janus-Kinase (JAK) family of receptor-associated tyrosine kinases. Activated JAKs recruit and activate signal transducer and activator of transcription (STATs), which in turn drive gene transcription.6 The specific JAK-STAT activated depends on the cytokine signal which includes the interferons (IFNs) and IFN-related cytokines, the common γ-chain cytokines, and the IL-6-type cytokines. Several studies have demonstrated a key role for JAK-STAT signalling in the pathogenesis of rheumatoid arthritis (RA).7 ,8 Previous studies have shown increased pSTAT3 and phosphoSTAT1 (pSTAT1) expression in Ps lesional skin9 and shown that IFNγ, IL-6 and IL-22 can induce pSTAT1 or pSTAT3 in keratinocytes;10 however, no study to date has examined their expression or regulation in PsA synovium or fibroblasts. Tofacitinib is a drug of the JAK inhibitor class and is currently approved for the treatment of RA in 27 countries. A recent study in patients with RA demonstrated that tofacitinib downregulated inflammatory pathways in RA synovial tissue;11 however, a direct effect of tofacitinib on PsA synovial inflammation has not been shown to date.
Methods
Patient demographics, arthroscopy and culture of synovial fibroblasts
See online supplementary file 1.
Synovial tissue explant culture ex vivo
To investigate the effect of tofacitinib (Selleck Chemicals LLC; Houston, Texas, USA) on cytokine production in the arthritic joint, an ex vivo PsA synovial explant model was established. This system maintains the synovial architecture and cell–cell contact and spontaneously releases proinflammatory mediators. PsA synovial biopsies were sectioned and cultured immediately following arthroscopy (to maintain maximal activity of the inflamed synovium) in 96-well plates (Falcon, Franklin Lakes, New Jersey, USA) in RPMI-1640 supplemented with streptomycin (100 units/mL) and penicillin (100 units/mL) for 24 h at 37°C in 5% CO2. Explants were cultured with 1 µM tofacitinib or dimethyl sulfoxide (DMSO) vehicle control for 72 h. Following culture, biopsy wet-weights were obtained and supernatants analysed for cytokines. Tissue morphology and cell viability of PsA explants following culture are described in online supplementary file 1.
Western blot analysis
Protein isolation from Psoriatic Arthritis synovial fibroblasts (PsAFLS) and synovial explants is described in online supplementary file 1. Protein (20–50 µg) was resolved on sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (5% stacking, 10% resolving), gels were then transferred onto nitrocellulose membranes (Amersham Biosciences, Buckinghamshire, UK) prior to 1 h blocking in wash buffer containing 5% non-fat milk. Membranes were incubated with rabbit polyclonal anti-pSTAT3 (Cell-Signaling Technology, UK), total-signal transducer and activator of transcription (tSTAT)3, pSTAT1, tSTAT1, pSTAT2, suppressor of cytokine signaling-3 (SOCS3), protein inhibitor of activated Stat3 (PIAS3; Cell Signaling Technology) and nuclear factor kappa B cells (NFκBp65) (Millipore, California, USA) diluted in 5% non-fat milk containing 0.1% Tween 20 at 4°C overnight. β-Actin (Sigma-Aldrich) was used as a loading control. Membranes were incubated with appropriate horseradish peroxidase-conjugated secondary antibodies for 3 h at RT. Signal was detected using SuperSignal West-Pico Chemiluminescent Substrate (Amersham Biosciences, UK) and quantified using EDAS-120 system (Kodak, Rochester, New York, USA).
PsAFLS invasion, migration, cytokine/matrix metallopeptidase quantification and statistics
See online supplementary file 1.
Results
Tofacitinib regulates STAT signalling in primary PsAFLS
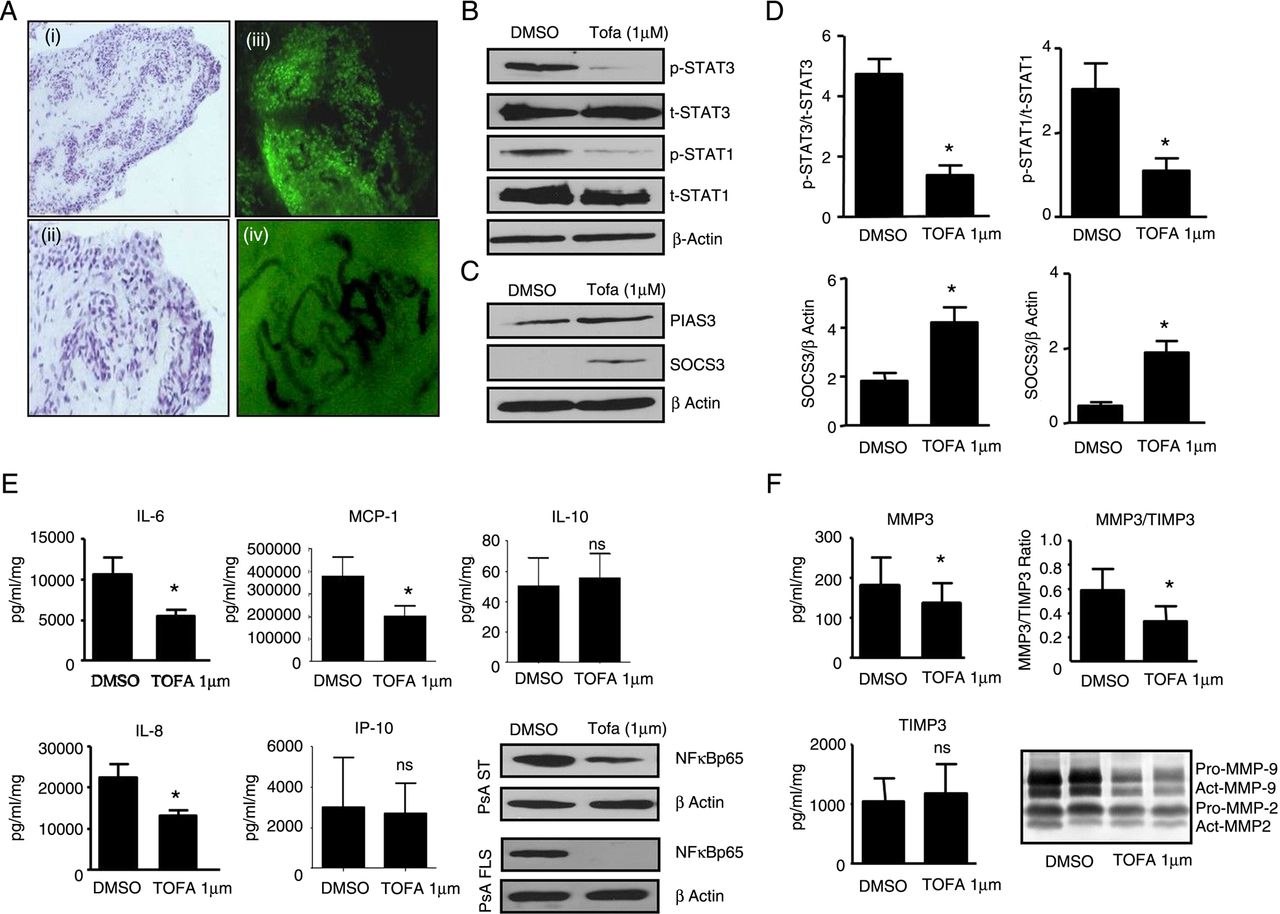
Initial experiments demonstrated increased expression of pSTAT1 and pSTAT3 in PsA synovium when compared with osteoarthritis (OA) (figure 1A). Tofacitinib inhibited pSTAT3 and pSTAT1 expression in PsAFLS when compared with vehicle control (figure 1B, D). In parallel, tofacitinib induced negative inhibitors of STATs, SOCS3 and PIAS3 compared with vehicle control (figure 1C, D).

Tofacitinib regulates Janus-kinase/signal transducer and activator of transcription (JAK/STAT) pathways components in PsAFLS. (A) Representative western blot showing phosphoSTAT3 (pSTAT3)/phosphoSTAT1 (pSTAT1) and tSTAT3/tSTAT1 protein in psoriatic arthritis (PsA) vs osteoarthritis (OA) synovial biopsy lysates. (B) Representative western blot showing pSTAT3/pSTAT1 and tSTAT3/tSTAT1 protein in dimethyl sulfoxide (DMSO) control vs tofacitinib (TOFA) (1 µM) treated PsA synovial fibroblasts (PsAFLS) for 24 h. Densitometry quantification of pSTAT3/pSTAT1 in PsAFLS (n=7). Data were normalised to tSTAT1/tSTAT3. (C) Representative western blot showing the expression of Protein inhibitor of activated Stat3 (PIAS3) and suppressor of cytokine signaling-3 (SOCS3) protein in DMSO control vs TOFA (1 µM) treated PsAFLS for 24 h. Densitometry quantification of PIAS3 and SOCS3 in PsAFLS (n=7). Data are expressed as mean±SEM. *p<0.05 vs DMSO control; β-actin was used as loading control.
Tofacitinib inhibited PsAFLS invasion and migration
Figure 2A shows representative images, demonstrating the inhibitory effect of tofacitinib on PsAFLS migration, in comparison with DMSO control where repopulation of wound margins was observed. Representative images of decreased PsAFLS invasion following culture with tofacitinib are shown in figure 2B, an effect that was significant compared with vehicle control (p<0.05; figure 2C). Furthermore, tofacitinib significantly inhibited PsAFLS network formation (figure 2D, E; p<0.05).

Tofacitinib inhibited PsAFLS invasion. Representative photomicrographs showing psoriatic arthritis (PsA) synovial fibroblasts (PsAFLS) migration (A) and invasion (B) following inhibition with TOFA (0.5 and 1 µM) for 24 h and 48 h, respectively, of n=5 experiments, (magnification ×20). Dimethyl sulfoxide (DMSO) was used as vehicle control. (C) Representative bar graph quantifying PsAFLS invasion following inhibition with TOFA (0.5 and 1 µM) for 48 h. Data are expressed as mean±SEM of n=5 replicate experiments. (D) Representative photomicrographs showing PsAFLS network formation following TOFA (0.5 and 1 µM) inhibition for 24 h compared with DMSO control. (E) Bar graph demonstrating the PsA network formation quantification following TOFA (0.5 and 1 µM) inhibition for 24 h. Data are expressed as mean±SEM of n=5 replicate experiments. *p<0.05 versus DMSO vehicle control.
Tofacitinib regulates STAT signalling in whole tissue PsA synovial explant cultures
To further examine the effect of tofacitinib, PsA synovial explants were cultured with tofacitinib for 72 h. Figure 3A(i–ii) demonstrates intact tissue morphology and cell–cell contact following culture, with lining-layer, blood vessels and synovial infiltrate clearly visible. Figure 3Ciii demonstrates calcein uptake into the nucleus of the cells following culture indicating viability of cells. Also evident are tortuous elongated dilated vessels which are a hallmark of PsA synovium (figure 3Aiv), with a mean (±SEM) vascularity of 75±5.3. Tofacitinib inhibited pSTAT1 and pSTAT3, with no effect on tSTAT1 or tSTAT3 compared with DMSO control (figure 3B, D), pSTAT2 was undetectable. In contrast, tofacitinib induced expression of SOCS3 and PIAS3 (figure 3C, D).

{kind=link}
{kind=link}
{kind=link}
Janus-kinase/signal transducer and activator of transcription (JAK/STAT) pathway components were modulated by tofacitinib in synovial tissues. (A) Representative images of psoriatic arthritis (PsA) explants following cultures. Histological images of explant tissue following culture at low (i) and high power (ii) showing intact tissue morphology and cell–cell contact with lining layer (black arrows), blood vessels (red arrows) and synovial infiltrate clearly visible. (iii) Calcein uptake into the nucleus of the cells in PsA whole mounts following culture indicating viability of cells, also evident are the tortuous elongated dilated vessels which are a hallmark of PsA synovium (iv). Representative western blot showing pSTAT3/pSTAT1 and tSTAT3/tSTAT1 protein in dimethyl sulfoxide (DMSO) control vs TOFA (1 µM) treated PsA synovial explants for 72 h (n=7; B). Representative western blot showing the expression of PIAS3 and suppressor of cytokine signaling-3 (SOCS3) protein DMSO control vs TOFA (1 µM) treated PsA synovial explants for 72 h (n=7; C). Densitometry quantification of phosphoSTAT3 (pSTAT3)/phosphoSTAT1 (pSTAT1) and protein inhibitor of activated Stat3 (PIAS3)/SOCS3 (D). Data were normalised to tSTAT3 or β-actin. Data are expressed as the mean±SEM of replicate experiments (n=7). *p<0.05 vs DMSO vehicle control; β-actin was used as loading control. TOFA (1 µM) inhibited spontaneous secretion of interleukin (IL)-6, IL-8, MCP-1, with no effect on IP-10 or IL-10 (E; n=7–11). Representative western blot showing inhibition of NFκBp65 in PsA synovial explants and PsA synovial fibroblasts (PsAFLS) following culture with TOFA (1 µM) compared with DMSO control (1 µM; E). β-actin was used as loading control. TOFA (1 µM) inhibited spontaneous secretion of MMP3, TIMP3 from PsA synovial explants and inhibited the MMP3/TIMP3 (F) ratio (n=6). DMSO was used as vehicle control. Representative in-gel zymography of MMP9/2 activities in PsA synovial explant cultured with TOFA (1 µM) for 72 h (F). DMSO was used as vehicle control (n=7). Data are expressed as mean±SEM. *p<0.05 vs DMSO control.
Inhibition of proinflammatory mediators from PsA explant cultures
Tofacitinib significantly inhibited spontaneous secretion of IL-6, IL-8 and monocyte chemoattractant protein-1 (MCP-1) from PsA explant cultures (*p<0.05; figure 3E) with no effect observed for IFN-gamma-inducible protein 10 (IP-10) or IL-10 (figure 3E). IL-17 levels were undetectable. Furthermore, tofacitinib inhibited NFκBp65 in PsA explants and PsAFLS, a key transcriptional factor in the inflamed joint (figure 3E). Tofacitinib significantly inhibited matrix metallopeptidase (MMP)3 expression from PsA synovial explants (p<0.05; figure 3F), had no effect on tissue inhibitor of metalloproteinases 3 (TIMP3) (figure 3F) and inhibited the MMP3/TIMP3 ratio (figure 3F). Finally tofacitinib inhibited MMP2 and MMP9 activity as observed by gelatine zymography (figure 3F).
Discussion
To date, no study has examined JAK-STAT or the effect of tofacitinib in primary cells isolated from PsA synovial biopsies. In this study, tofacitinib significantly decreased pSTAT1, pSTAT3 in PsAFLS and PsA synovial explant cultures ex vivo. In parallel, tofacitinib increased SOCS3 and PIAS3 expression demonstrating negative feedback inhibition. Functionally, tofacitinib significantly decreased PsAFLS invasion, migration and network formation. Finally, tofacitinib significantly decreased spontaneous secretion of key proinflammatory cytokines, the MMP/TIMP ratio and NFκBp65 expression. Thus tofacitinib inhibits proinflammatory and invasive mechanisms which are critically involved in the pathogenesis of PsA.
This is the first study to demonstrate regulation of pSTAT3 and pSTAT1 in primary PsAFLS and PsA synovial tissue. Only one previous study in PsA has examined STAT signalling, and this was in synovial fluid (SF) T cells where increased expression of JAK1, pSTAT3 and pSTAT1 was demonstrated compared with peripheral blood (PB) of healthy controls (HC), suggesting activation of JAK-STAT signalling at the site of inflammation.12 Ps studies have shown that increased pSTAT expression is associated with epidermal hyper-proliferation, expression of which was modulated by IFNγ, IL-6 and IL-22.9 ,10 This is consistent with RA studies, showing that pSTAT3 is associated with synovial inflammation, lining-layer hyperplasia and synovial pO2 levels,13 ,14 and that STAT3 modulates Th17 differentiation in RA SF and induces RAFLS survival.13
In this study, tofacitinib inhibited pSTAT1, pSTAT3 and NFκBp65 in PsAFLS and PsA explants, an effect that was paralleled by induction of both SOCS3 and PIAS3. SOCS inhibits cytokine signalling by acting as kinase inhibitors to JAKs or competitive binding for docking sites with STAT. PIAS3 binds to the STAT3 DNA-binding domains and thus prevents physical binding of STAT to target genes thus inhibiting transcriptional activity. In parallel, tofacitinib inhibited PsAFLS invasion, migration and network formation, all associated with progressive and destructive joint disease. While the precise mechanisms by which tofacitinib inhibits invasion/migration is unclear, it may involve (i) inhibition of key cytokines such as IL-6 which results in negative feedback inhibition, (ii) blockade of RhoGTPases (cdc42, Rac1 and RhoA) or growth factors such as platelet-derived growth factor (PDGF), which are critical for cell movement, migration and invasion or (iii) through the observed effects of STAT signalling on NFκB which is known in other cell types to mediate proliferation and invasive mechanisms via phosphoinositide 3-kinase (PI3K)/serine/threonine kinase or protein kinase B (AKT) signalling pathway.15
Tofacitinib significantly inhibited IL-6, IL-8, MCP-1, MMP3 and MMP2/9 spontaneous secretion from PsA explants, with no significant effect on IP-10 or IL-10. The PsA biopsies used to establish the explant model were obtained from the site of inflammation under direct visualisation and closely reflect the patients’ inflammatory activity in vivo. Previous studies have shown that tofacitinib inhibits tumour necrosis factor (TNF)-α and IL-6-induced osteoclastogenesis and bone destruction,16 an effect mediated by receptor activator of nuclear factor kappa-B ligand (RANKL).17 Tofacitinib decreases the T-cell stimulatory capability of dendritic cells through suppression of type-I-IFN signalling.18 Furthermore, tofacitinib reduces MMP and IFN-regulated gene expression in RA synovium, with clinical improvement correlating with reductions in pSTAT1 and pSTAT3.8 While JAK1-mediated IFN and IL-6 signalling likely play a key role in the synovial response, more recent data have shown that CP690550 inhibits TNF-induced expression of IP10, RANTES and MCP1 in RAFLS,11 inhibits IL-4-dependent Th2 cell differentiation and Th17 cell differentiation and decreases cartilage destruction through suppression of IL-17 and IFNγ-producing CD4+ T cells.19
In conclusion, this is the first study to demonstrate the effect of tofacitinib in PsAFLS and PsA explant cultures. Tofacitinib differentially regulated JAK-STAT signalling, inhibiting pSTAT1, pSTAT3 and inducing of SOCS3/PIAS3 in vitro and ex vivo paralleled by inhibition of invasive mechanisms. These data further support a role for blockade of JAK-STAT signalling pathways in the treatment strategy for PsA.
Acknowledgments
This work was supported by the Health Research Board, Programme for Research in Third Level Institutions (PRTLI), Ireland and the JU IMI BeTheCure award.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
GW and TMcG are joint first authors.
Contributors All authors have made substantial contributions to the conception or design of the work, or the acquisition, analysis or interpretation of data. All authors were involved in drafting the work or revising it critically for important intellectual content. All authors have read final approval of the version published.
Competing interests None declared.
Ethics approval St Vincent's University Hospital Ethics Committee.
Provenance and peer review Not commissioned; externally peer reviewed.