Article Text

Abstract
Objective Hypoxia-inducible factor 2α (HIF-2α), encoded by Epas1, causes osteoarthritic cartilage destruction by regulating the expression of matrix-degrading enzymes. We undertook this study to explore the role of nicotinamide phosphoribosyltransferase (NAMPT or visfatin) in HIF-2α-mediated osteoarthritic cartilage destruction.
Methods The expression of HIF-2α, NAMPT and matrix-degrading enzymes was determined at the mRNA and protein levels in human osteoarthritis (OA) cartilage, mouse experimental OA cartilage and primary cultured mouse chondrocytes. Experimental OA in mice was induced by destabilisation of the medial meniscus (DMM) surgery or intra-articular injection of Ad-Epas1 or Ad-Nampt in wild-type, Epas1+/−, Epas1fl/fl;Col2a1-Cre and Col2a1-Nampt transgenic (TG) mice. Primary cultured mouse chondrocytes were treated with recombinant NAMPT protein or were infected with adenoviruses.
Results We found that the Nampt gene is a direct target of HIF-2α in articular chondrocytes and OA cartilage. NAMPT protein, in turn, increased mRNA levels and activities of MMP3, MMP12 and MMP13 in chondrocytes, an action that was necessary for HIF-2α-induced expression of catabolic enzymes. Gain-of-function studies (intra-articular injection of Ad-Nampt; Col2a1-Nampt TG mice) and loss-of-function studies (intra-articular injection of the NAMPT inhibitor FK866) demonstrated that NAMPT is an essential catabolic regulator of osteoarthritic cartilage destruction caused by HIF-2α or DMM surgery.
Conclusions Our findings indicate that NAMPT, whose corresponding gene is a direct target of HIF-2α, plays an essential catabolic role in OA pathogenesis and acts as a crucial mediator of osteoarthritic cartilage destruction caused by HIF-2α or DMM surgery.
- Knee Osteoarthritis
- Osteoarthritis
- Chondrocytes
- Cytokines
- Synovitis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Osteoarthritis (OA) is a degenerative joint disease that is primarily characterised by cartilage destruction. A variety of aetiological risk factors and pathophysiological processes contribute to the progressive nature of the disease. Important among potential OA-causing mechanisms are mechanical stresses, including joint instability and injury, and factors that predispose toward OA, such as ageing. These factors lead to the activation of biochemical pathways in chondrocytes that result in degradation of the extracellular matrix (ECM) by matrix metalloproteinases (MMPs) and aggrecanases (ADAMTS).1–3 We have previously demonstrated that hypoxia-inducible factor (HIF)-2α, encoded by EPAS1, triggers osteoarthritic cartilage destruction by upregulating chondrocyte expression of matrix-degrading enzymes.4–6
In a preliminary study, we identified Nampt as a HIF-2α-upregulated gene in mouse chondrocytes. Nampt encodes nicotinamide phosphoribosyltransferase, which functions as both an intracellular form (iNAMPT) and an extracellular form (eNAMPT). eNAMPT, also known as visfatin, acts as an adipokine, whereas the nicotinamide phosphoribosyltransferase enzymatic activity of iNAMPT regulates salvage pathways of NAD+ synthesis.7 ,8 eNAMPT displays pro-inflammatory functions and appears to be associated with inflammatory diseases, such as rheumatoid arthritis (RA).9 ,10 For instance, levels of eNAMPT in the synovial fluid of RA patients are positively related to disease severity.11–13 eNAMPT regulates the expression of chemokines and cytokines in cell types involved in RA pathogenesis.14 ,15 Additionally, inhibition of iNAMPT enzymatic activity with FK86616 blocks experimental RA.17 ,18 Similar to their association with RA, elevated eNAMPT levels are positively linked to OA pathogenesis.19 eNAMPT in human OA chondrocytes inhibits proteoglycan synthesis and increases the expression of matrix-degrading enzymes.20 ,21 Additionally, interleukin (IL)-1β acts through overexpression of NAMPT to inhibit collagen type II (COL2A1) expression in chondrocytes.22 Although these observations suggest a possible role for NAMPT in OA pathogenesis, the contribution of NAMPT in OA pathogenesis in vivo remains to be established. In this study, we investigated the role and underlying molecular mechanisms of HIF-2α-upregulated NAMPT in OA pathogenesis.
Materials and methods
OA cartilage and experimental OA
Human OA cartilage was sourced from individuals undergoing arthroplasty.4–6 Male C57BL/6, STR/ort, CBA/CaCrl, Col2a1-Nampt transgenic (TG), Epas1+/−, Epas1fl/fl;Col2a1-Cre and Il6−/− mice were used for experimental OA studies. Col2a1-Nampt TG mice were generated using the Col2a1 promoter and enhancer.4–6 Epas1+/−, Epas1fl/fl and Col2a1-Cre TG mice were obtained from the Jackson Laboratory. Epas1fl/fl;Col2a1-Cre and Il6−/− mice were described previously.5 ,6 Spontaneous OA in STR/ort23 and Nampt TG mice was examined at 28 and 55 weeks of age, respectively. Experimental OA was induced by destabilisation of the medial meniscus (DMM) surgery. Mice were also injected intraperitoneally with FK886 (10 mg/kg) once every 3 days and sacrificed 8 weeks after DMM surgery. Experimental OA was additionally induced by intra-articular injection (once weekly for 3 weeks) of Ad-Nampt or Ad-Epas1 (1×109 plaque forming units) with or without FK886. Mice were sacrificed 21 days after the first intra-articular injection.4–6 ,24
Histology and immunohistochemistry
Human OA cartilage sections were stained with alcian blue. Cartilage destruction in mice was examined using safranin-O staining and scored using the OARSI grading system.25 Synovitis was determined by haematoxylin staining. Detailed procedures are presented in online supplementary materials and methods.
Other materials and methods
Additional methods are described in online supplementary materials and methods. These include histology and immunohistochemistry; primary culture of articular chondrocytes; microarray analysis; adenoviruses, infection of chondrocytes and intra-articular injection of mice; ELISA; reverse transcription-PCR (RT-PCR), quantitative RT-PCR (qRT-PCR) and small inhibitory RNA; western blotting; cloning, reporter gene assays and ChIP assays; skeletal staining; and MMP activity assays. PCR primer and siRNA sequences are summarised in online supplementary Table S1 and Table S2, respectively.
Statistical analysis
Values are presented as means±SEM. The n numbers indicated for each figure correspond to the number of independent experiments or the number of mice used. Data quantified based on an ordinal grading system, such as OARSI grade, were analysed using non-parametric statistical methods. For qRT-PCR, data expressed as relative fold changes, Student t test and analysis of variance with post hoc tests were used for pair-wise comparisons and multi-comparisons, respectively, after first confirming a normal distribution using the Shapiro–Wilk test. Significance was accepted at the 0.05 level of probability (p<0.05).
Results
HIF-2α directly targets the Nampt gene in articular chondrocytes
Microarray analyses of chondrocytes infected with Ad-Epas1 indicated upregulation of Nampt (visfatin) and downregulation of Adipoq (adiponectin), Retn (resistin) and Lep (leptin) (online supplementary figure S1). Differential adipokine expression was additionally confirmed in primary cultured chondrocytes (figure 1A). We focused on the role of NAMPT, whose corresponding gene is positively regulated by HIF-2α, in OA pathogenesis. ChIP assays revealed that HIF-2α binds to the proximal site among the three HIF-2α binding sites (CGTG) in the mouse Nampt promoter (figure 1B). HIF-2α stimulated Nampt promoter activity, an effect that was inhibited by co-transfection of dominant-negative (Δ) HIF-2α (figure 1C). Mutation of the proximal site, but not other HIF-2α binding sites, inhibited reporter gene activity (figure 1C). These results collectively show that the Nampt gene is a direct target of HIF-2α in chondrocytes.
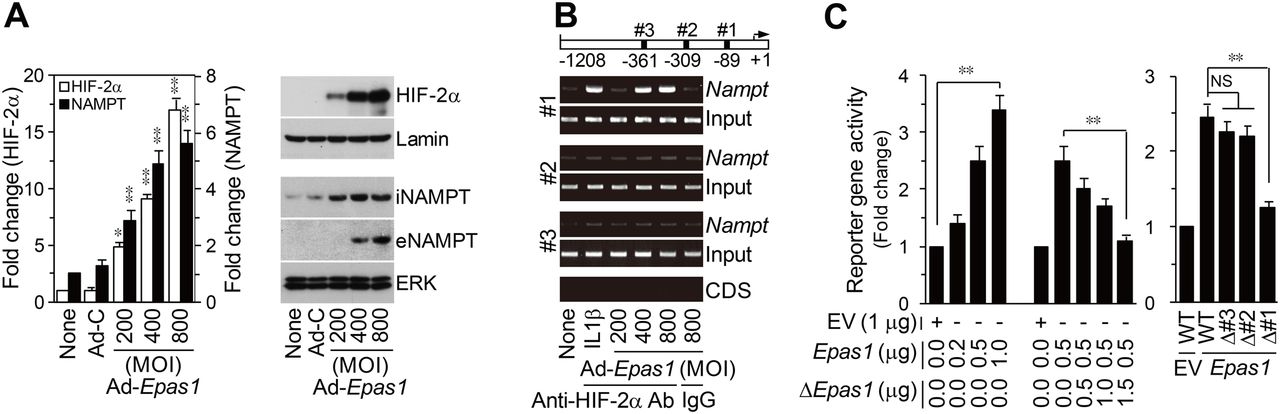
Nampt is a direct target gene of HIF-2α in mouse articular chondrocytes. (A) qRT-PCR (n=6) and western blotting of HIF-2α and NAMPT in chondrocytes infected with Ad-C (800 MOI) or Ad-Epas1 for 24 h. (B) HIF-2α binding to the Nampt promoter was detected by ChIP assay in chondrocytes treated with IL-1β (1 ng/mL) or infected with Ad-Epas1. The Nampt coding sequence (CDS) was used as a negative control. IgG was used as an isotype control for the anti-HIF-2α antibody. (C) Nampt reporter gene activity in chondrocytes transfected with empty vector (EV), Epas1 vector or ΔEpas1 vector (n=6) (left). Reporter gene activity of wild-type (WT) and HIF-2α binding site-mutated reporter genes (n=6) (right). *p<0.01, **p<0.001.
NAMPT is required for HIF-2α-induced MMP expression
We next investigated the possible role of NAMPT in the expression of matrix-degrading enzymes and ECM proteins in chondrocytes. Overexpression of NAMPT by Ad-Nampt infection induced upregulation and activation of the MMP3, MMP12 and MMP13, and downregulation of aggrecan without significant effects on COL2A1 expression (figure 2A,B and online supplementary figure S2A). ELISAs revealed that both iNAMPT and eNAMPT were produced by Ad-Nampt infection (figure 2C). The significance of NAMPT enzymatic activity was determined using the specific inhibitor FK866.16 FK866 significantly blocked Ad-Nampt-induced upregulation of MMP, MMP12 and MMP13 (figure 2D and online supplementary figure S2B). eNAMPT action was assessed by treating chondrocytes with recombinant mouse NAMPT protein (rNAMPT). rNAMPT stimulated the expression of MMPs (MMP3, MMP12 and MMP13) and downregulated aggrecan (figure 2E and online supplementary figure S2C). However, addition of a neutralising antibody against eNAMPT did not affect Ad-Nampt-induced upregulation of MMP3, MMP12 and MMP13 (online supplementary figure S2D). Additionally, HIF-2α-induced MMP expression was significantly blocked by knockdown or inhibition of NAMPT (figure 2F and online supplementary figure S2E). NAMPT overexpression in chondrocytes also upregulated various cytokines and chemokines (online supplementary figure S3). Among these, IL-6 induced upregulation of MMP3 and MMP13, consistent with a previous report.6 Experiments using chondrocytes from Il6−/− mice demonstrated that NAMPT-induced MMP3 expression was at least partly mediated by IL-6 production (online supplementary figure S3). However, treatment of Ad-Epas1-infected chondrocytes with other adipokines (ie, resistin, adiponectin and leptin) did not affect HIF-2α-induced upregulation of MMPs (online supplementary figure S4). Collectively, these results indicate that NAMPT promotes upregulation of MMP3, MMP12 and MMP13 in chondrocytes, an action that is necessary for HIF-2α-induced expression of catabolic enzymes.

NAMPT is required for HIF-2α-induced matrix metalloproteinase (MMP) expression in chondrocytes. (A) Detection of the indicated mRNAs/proteins by qRT-PCR/western blotting in chondrocytes infected with Ad-C or Ad-Nampt (n=6). (B) MMP activity assays in chondrocytes infected with Ad-C (800 MOI) or Ad-Nampt (n=5). (C) NAMPT levels quantified by ELISA (n=5) in chondrocytes infected with Ad-C (800 MOI) or Ad-Nampt. (D) Expression levels of catabolic factors determined by qRT-PCR (n=6) in chondrocytes infected at an MOI of 800 with Ad-C or Ad-Nampt in the absence or presence of FK866 (100 μM). (E) qRT-PCR (n=6) in chondrocytes treated with recombinant NAMPT (rNAMPT). (F) Chondrocytes were transfected with 100 nM control siRNA (C-siRNA) or Nampt siRNA, or were treated with or without FK866 (100 μM) and exposed to Ad-Epas1 for 36 h. mRNA levels were determined by qRT-PCR (n=6). *p<0.01, **p<0.001.
NAMPT is upregulated in OA cartilage
To ascertain whether NAMPT plays a role in OA pathogenesis, we examined NAMPT expression in OA cartilage of humans and mice. NAMPT mRNA and protein were markedly elevated in OA-affected human cartilage compared with undamaged regions of arthritic cartilage (figure 3A). NAMPT levels were also increased in mouse OA models, including STR/ort mouse OA cartilage (figure 3B) and OA cartilage induced by DMM surgery (figure 3C). OA cartilage in both humans and mouse models shows increased expression of HIF-2α.4–6 Consistent with this, overexpression of HIF-2α in articular cartilage by intra-articular injection of Ad-Epas1 increased NAMPT levels, with concomitant osteoarthritic cartilage destruction (figure 4A). Additionally, DMM-induced NAMPT upregulation and OA cartilage destruction were significantly blocked in chondrocyte-specific Epas1 conditional knockout (CKO) mice (figure 4B,C), indicating that NAMPT expression in chondrocytes of OA cartilage is regulated by HIF-2α.

NAMPT is overexpressed in human and mouse osteoarthritis (OA) chondrocytes. (A) Alcian blue staining, NAMPT immunostaining and NAMPT mRNA levels quantified by qRT-PCR of human OA cartilage. (B and C) Spontaneous OA cartilage from STR/ort mice and CBA control mice (B), and sham- and destabilisation of the medial meniscus (DMM)-operated C57BL/6 mice (C). mRNA levels in cartilage tissue were quantified by qRT-PCR (n=10). **p<0.001. Scale bar: 50 μm.
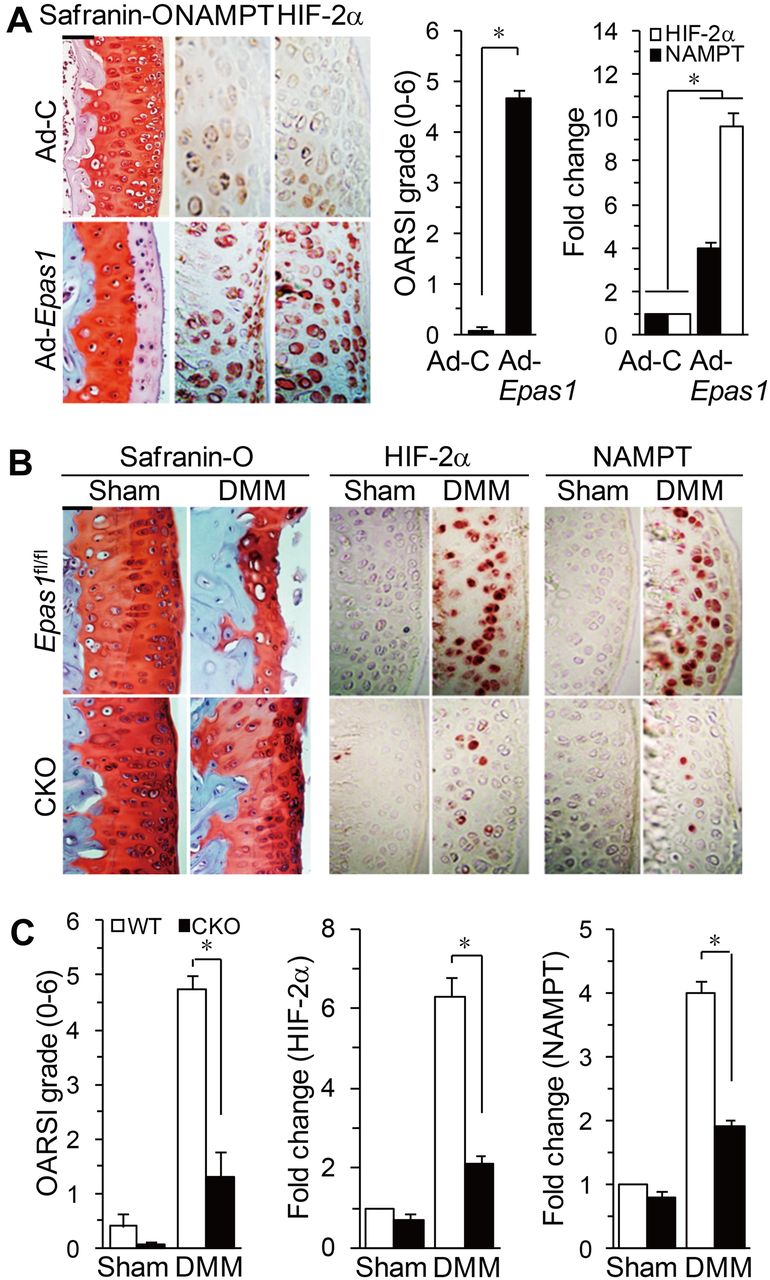
NAMPT expression in osteoarthritis cartilage is regulated by HIF-2α. (A) Cartilage tissue of mice, intra-articularly injected with Ad-C or Ad-Epas1, was used for safranin-O staining, OARSI grade, and immunodetection/qRT-PCR of HIF-2α and NAMPT proteins/mRNAs (n=6). (B and C) Safranin-O staining, immunostaining for HIF-2α and NAMPT, OARSI grade, and qRT-PCR of NAMPT and HIF-2α mRNAs (n=10) in wild-type (WT) and Epas1 conditional knockout (CKO) mice following destabilisation of the medial meniscus (DMM) surgery. *p<0.001. Scale bar: 100 μm.
NAMPT causes experimental OA cartilage destruction
The role of NAMPT in OA pathogenesis in mice was examined by intra-articular injection of Ad-Nampt, which caused NAMPT overexpression in cartilage, meniscus and synovium (figure 5A and online supplementary figure S5A). Gene delivery by intra-articular injection of adenovirus was further confirmed by monitoring injected Ad-Gfp (online supplementary figure S5B). Ad-Nampt injection triggered cartilage destruction with significantly increased expression of the NAMPT targets MMP3, MMP12 and MMP13 in cartilage tissue (figures 5A,B). Ad-Nampt injection additionally resulted in synovitis (online supplementary figure S5C). The role of NAMPT was further validated by generating Col2a1-Nampt TG mice. Chondrocytes from these mice, which showed no developmental defects (online supplementary figure S6), exhibited elevated mRNA levels of NAMPT and its target genes compared with wild-type (WT) chondrocytes (figure 5C). Consistent with this, Col2a1-Nampt TG mice showed significantly enhanced osteoarthritic cartilage destruction following DMM surgery (figure 5D). Moreover, aged (12-month-old) TG mice showed spontaneous cartilage destruction compared with WT mice of the same age (figure 5E). Thus, gain-of-function studies of NAMPT collectively indicate that NAMPT is an important catabolic regulator of OA pathogenesis.

Overexpression of NAMPT in chondrocytes causes osteoarthritis cartilage destruction. (A and B) Mice were Intra-articularly injected with Ad-C or Ad-Nampt and evaluated by (A) NAMPT immunostaining, safranin-O staining and OARSI grading (n=6), and (B) qRT-PCR (n=6) of catabolic factors in cartilage tissues. (C) qRT-PCR of mRNAs in chondrocytes isolated from wild-type (WT) or Col2a1-Nampt transgenic (TG) mice (n=7). (D) NAMPT immunostaining, safranin-O staining and OARSI grade (n=13) in destabilisation of the medial meniscus (DMM)- or sham-operated WT and Col2a1-Nampt TG mice. (E) Spontaneous cartilage destruction in 12-month-old WT and TG mice determined by safranin-O staining and OARSI grade (n=10). *p<0.01, **p<0.001. Scale bar: 100 μm.
NAMPT is required for experimental OA cartilage destruction
Because iNAMPT enzymatic activity is necessary for MMP expression, we examined whether iNAMPT regulates OA pathogenesis. Injection of FK866 (intraperitoneally or intra-articularly) significantly inhibited cartilage destruction induced by Ad-Nampt injection (figure 6A), with concomitant reduction of the mRNA levels NAMPT targets MMP3, MMP12 and MMP13 in cartilage tissue (online supplementary figure S7A). Intraperitoneal injection of FK866 also inhibited DMM-induced cartilage destruction (figure 6B) and upregulation of NAMPT target MMPs (online supplementary figure S7B). Finally, injection of FK866 (intraperitoneally or intra-articularly) blocked Ad-Epas1-induced cartilage destruction and expression of NAMPT target MMPs (figure 6C and online supplementary figure S7C). Additionally, synovitis caused by Ad-Epas1 or Ad-Nampt injection was blocked by co-injection of FK866 (online supplementary figure S8). However, knockdown of Epas1 in Epas1+/− or Epas1fl/fl;Col2a1-Cre mice did not affect the cartilage destruction induced by Ad-Nampt injection (online supplementary figure S9), suggesting that NAMPT is downstream of HIF-2α. These results collectively indicate that iNAMPT enzymatic activity is necessary for OA pathogenesis induced by DMM surgery or intra-articular injection of Ad-Nampt or Ad-Epas1.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Inhibition of NAMPT enzymatic activity blocks osteoarthritis cartilage destruction. (A) Mice were intra-articularly injected with Ad-C or Ad-Nampt and co-injected (intra-articularly or intraperitoneally) with FK866. Cartilage destruction was detected by safranin-O staining and quantified by OARSI grade (n=10). (B) Mice were intraperitoneally injected with FK866 after destabilisation of the medial meniscus (DMM) surgery. Cartilage destruction was detected and quantified (n=10). (C) Mice were intra-articularly injected with Ad-C or Ad-Epas1 and co-injected (intra-articularly or intraperitoneally) with FK866. Cartilage destruction was detected and quantified (n=10). **p<0.001. Scale bar: 100 μm.
Discussion
Catabolic signals in OA pathogenesis are categorised into two main pathways: upregulation of tissue-destructive enzymes such as MMP3, MMP13 and ADAMTS5, and downregulation of cartilage-specific ECM proteins such as type II collagen and aggrecan. HIF-2α serves as an essential catabolic regulator of OA pathogenesis by upregulating tissue-destructive genes.4–6 We demonstrated here that NAMPT, whose corresponding gene is a direct target of HIF-2α, plays an essential role in OA pathogenesis by upregulating matrix-degrading enzymes and downregulating aggrecan expression. Our gain-of-function studies (intra-articular injection of Ad-Nampt; Col2a1-Nampt TG mice) and loss-of-function studies (intra-articular injection of FK866) demonstrated that NAMPT is an essential catabolic regulator of OA pathogenesis. NAMPT appears to exert its catabolic functions by downregulating aggrecan and upregulating MMP3 and MMP13, which are critical effectors of OA pathogenesis.26 ,27 The mechanism by which NAMPT regulates MMP expression was not elucidated in this study. However, because NAMPT acts as an enzyme and adipokine, we speculate that NAMPT regulates the expression of MMP genes indirectly, rather than by directly modulating the activity of their promoters. Possible mechanisms include modulation of chromatin functions by NAMPT-regulated members of the Sirtuin family7 ,28 and regulation by secondary mediators such as cytokines and chemokines that are upregulated by NAMPT. Indeed, we found that NAMPT-induced MMP3 expression is mediated, at least in part, by IL-6. In addition to the NAMPT target MMPs, MMP9 was also upregulated in OA cartilage induced in response to DMM surgery or Ad-Epas1 injection, and intra-articular injection of FK866 inhibited both cartilage destruction and MMP9 expression. Because NAMPT does not directly modulate MMP9 expression, we speculate that the inhibitory effect of FK866 on MMP9 expression is attributable to inhibition of osteoarthritic cartilage destruction by FK866.
Our results suggest that iNAMPT enzymatic activity plays an important role in the regulation of catabolic factor expression and is sufficient to promote OA pathogenesis. Consistent with this, administration of FK866 abolished experimental OA induced by DMM surgery or injection of Ad-Nampt or Ad-Epas1. However, eNAMPT also appears to plays a role in OA pathogenesis. This is clearly demonstrated by the observations that Ad-Nampt or Ad-Epas1 infection increased both iNAMPT and eNAMPT and that rNAMPT treatment caused upregulation of MMP3, MMP12 and MMP13. To date, the contribution of NAMPT to OA pathogenesis has not been established in vivo, although in vitro studies have suggested possible functions in OA pathogenesis.19–22 Therefore, our results are the first evidence for in vivo catabolic functions of NAMPT in the pathogenesis of OA. Intra-articular injection of Ad-Nampt also caused synovitis, which was blocked by inhibition of NAMPT enzymatic activity. Because OA is a disease that involves all joint tissues, including cartilage, synovium and subchondral bone, the synovial inflammation may affect cartilage destruction; hence, the inhibitory effects of the NAMPT inhibitor FK866 on synovitis may contribute to the observed effects on cartilage destruction.
NAMPT enzymatic activity regulates salvage pathways of NAD+ synthesis,7 ,8 and NAD+, in turn, is an essential cofactor of Sirtuin (SIRT) family protein deacetylases.7 ,29 Indeed, we found that NAMPT and HIF-2α increased NAD+ levels and SIRT1 activity in chondrocytes (data not shown). Among Sirtuin family members, SIRT1 is known to deacetylate HIF-2α,30 ,31 whereas SIRT7 negatively regulates HIF-2α protein stability.32 Evidence suggests a possible function of SIRT1 in OA pathogenesis through the regulation of cartilage ECM expression and chondrocyte apoptosis.33 ,34 Additionally, Sirt1-CKO mice or Sirt1 mutant mice exhibit enhanced osteoarthritic cartilage destruction,34 ,35 suggesting that SIRT1 plays a cartilage-protective role. This is a somewhat contradictory finding given the pro-inflammatory and catabolic functions of its upstream activator NAMPT. However, we found that SIRT activity is required for protein stability and transcriptional activity of HIF-2α, and thereby regulates the induction of matrix-degrading enzymes and OA pathogenesis in various mouse models (data not shown). This suggests that other member(s) of the Sirtuin family, rather than SIRT1, is associated with HIF-2α/NAMPT regulation of OA pathogenesis.
In summary, we found that Nampt is a direct target gene of HIF-2α in articular chondrocytes and is upregulated in OA cartilage. We also demonstrated by gain-of-function/loss-of-function studies that NAMPT plays an essential catabolic role in OA pathogenesis and acts as a crucial mediator of osteoarthritic cartilage destruction caused by HIF-2α or DMM surgery. Therefore, NAMPT could be an effective therapeutic target in the treatment of OA disease. Indeed, inhibition of NAMPT enzymatic activity by injection of FK866 (intra-articularly or intraperitoneally) inhibits osteoarthritic cartilage destruction caused by DMM surgery or intra-articular injection of Ad-Epas1 or Ad-Nampt.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Handling editor Tore K Kvien
-
SY and J-HR contributed equally.
-
Contributors SY designed and performed most of the in vitro and in vivo studies. J-HR conceived the project and carried out experiments. HO, JJ, J-SK and J-HK performed experimental work and analysed the data. HAK provided reagents and discussed the manuscript. C-HC provided and evaluated human joint samples. J-SC conceived the project and was responsible for the overall design and oversight of the project.
-
Funding This work was supported by grants from the National Research Foundation of Korea (2007-0056157, 2012M3A9B44028559 and 2013R1A2A1A01009713) and the Korea Healthcare Technology R&D project (A110274).
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval The Institutional Review Board of the Wonkwang University Hospital approved the use of human materials. All animal experiments were approved by the Gwangju Institute of Science and Technology Animal Care and Use Committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.