Article Text

Abstract
Objective: To characterise the expression of matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs) during degeneration of articular cartilage in a transgenic Del1 mouse model for osteoarthritis.
Methods: Northern analysis was used to measure mRNA levels of MMP-2, -3, -8, -9, -13, and -14, and TIMP-1, -2, and -3 in total RNA extracted from knee joints of transgenic Del1 mice, harbouring a 15 amino acid deletion in the triple helical domain of the α1(II) collagen chain, using their non-transgenic littermates as controls. Immunohistochemistry was used to study the presence of cleavage products (neoepitopes) of type II collagen, and the distribution of MMP-13 and TIMP-1 in degenerating cartilage.
Results: Each of the MMP and TIMP mRNAs analysed exhibited distinct expression patterns during development and osteoarthritic degeneration of the knee joint. The most striking change was up regulation of MMP-13 mRNA expression in the knee joints of Del1 mice at the onset of cartilage degeneration. However, the strongest immunostaining for MMP-13 and its inhibitor TIMP-1 was not seen in the degenerating articular cartilage but in synovial tissue, deep calcified cartilage, and subchondral bone. The localisation of type II collagen neoepitopes in chondrocytes and their pericellular matrix followed a similar pattern; they were not seen in cartilage fibrillations, but in adjacent unaffected cartilage.
Conclusion: The primary localisation of MMP-13 and TIMP-1 in hyperplastic synovial tissue, subchondral bone, and calcified cartilage suggests that up regulation of MMP-13 expression during early degeneration of articular cartilage is a secondary response to cartilage erosion. This interpretation is supported by the distribution of type II collagen neoepitopes. Synovial production of MMP-13 may be related to removal of tissue debris released from articular cartilage. In the deep calcified cartilage and adjacent subchondral bone, MMP-13 probably participates in tissue remodelling.
- osteoarthritis
- transgenic mice
- matrix metalloproteinases
- type II collagen
- MMPs, matrix metalloproteinases
- OA, osteoarthritis
- PBS, phosphate buffered saline
- RT-PCR, reverse transcription-polymerase chain reaction
- TIMP, tissue inhibitor of metalloproteinases
Statistics from Altmetric.com
- MMPs, matrix metalloproteinases
- OA, osteoarthritis
- PBS, phosphate buffered saline
- RT-PCR, reverse transcription-polymerase chain reaction
- TIMP, tissue inhibitor of metalloproteinases
Non-reversible damage of articular cartilage is a key feature in the pathogenesis of degenerative joint disease or osteoarthritis (OA). Despite intensive research, the pathogenetic mechanisms which result in gradual degeneration of articular cartilage, especially in weightbearing areas of joints, remain poorly understood.1,2 For example, little is known of the respective roles of physical trauma and enzymatic degradation of articular cartilage in the development of OA lesions. Arguments favouring the role of physical destruction of articular cartilage stem from the observation that acute trauma and experimental surgical defects cause damage that rarely undergoes complete healing, but results in progressive OA degeneration.3,4 If the defect extends into subchondral bone, better results can be expected through the activity of chondrogenic progenitors derived from bone marrow. Even under these circumstances the repair tissue is largely fibrocartilaginous in nature and has inferior structural properties.3–5 In adulthood, synthesis of collagen, especially of type II collagen by articular chondrocytes, is markedly reduced, which probably results in gradual weakening of the tissue.6,7 We have recently shown that adult articular chondrocytes can reactivate collagen production, but only to a limited amount, which is not sufficient to repair severe cartilage defects.8
There are also arguments favouring the role of proteolytic enzymes, especially those capable of degrading the collagen network, in the development of osteoarthritic cartilage damage. In many cases cartilage fibrillation is associated with superficial loss of proteoglycans, resulting in slowly progressing erosion of articular cartilage and exposure of the underlying subchondral bone.1,2 Destruction of the collagen network due to an altered balance of proteolytic enzymes, especially matrix metalloproteinases (MMPs), and their natural inhibitors, tissue inhibitors of metalloproteinases (TIMPs),9,10 may explain this type of progression. The MMP family currently consists of more than 20 zinc dependent, neutral endopeptidases.4,9 Evidence for degradation of cartilage collagen fibrils by interstitial collagenases comes from immunodetection of neoepitopes in OA cartilage created in type II collagen at two specific cleavage sites.11,12 Analogously, increased activities of interstitial collagenases MMP-1 and MMP-13 have been reported in human osteoarthritic cartilage and in experimental animal models for OA.13,14 Increased MMP-13 expression is not specific to the OA joints, as similar changes have been seen also in rheumatoid arthritis,15–17 and in healing traumas of articular cartilage.18 Synovial expression of MMP-13 makes it a potentially important enzyme in the pathogenesis of both arthritides.15 Additionally, it has also been suggested that two gelatinases, MMP-2 and MMP-9, and a membrane bound collagenase, MMP-14, play a part in matrix degradation.17,19,20
The metabolism of MMPs is tightly regulated, intracellularly at the level of transcription, translation, and secretion, and extracellularly by zymogen activation and inhibition by TIMPs,10,21 a family of four structurally related polypeptides.9,10 Synthesis of TIMP-1 and TIMP-2 has been detected in normal articular cartilage, whereas synovial TIMP-1 production has only been reported in OA joints.22–24
We have recently described a Del1 mouse model for OA, in which a deletion mutation in the type II collagen transgene results in a structurally inferior collagen network and predisposes the animals to early onset OA of the knee joints.7,25 Articular cartilage erosion is started by superficial fibrillation at the age of 3 months. Simultaneously with the onset of cartilage erosion, increased synthesis of cartilage oligomeric matrix protein, and its secretion into serum is seen.26 Histologically the disease progresses to severe erosion of articular cartilage at central condylar areas, reaching the tidemark by 6 months and subchondral bone by 9 to 15 months of age. The entire joint is affected in the degenerative process that includes subchondral bone remodelling, mineralisation of ligaments and tendon, and severe degeneration of menisci.7 Although inferior physical properties of cartilage form the most likely background to the OA seen in Del1 mice, the disease process thus also involves extensive tissue remodelling, suggesting involvement of MMPs and TIMPs. We therefore decided to characterise the production of MMPs and TIMPs and of type II collagen derived neoepitopes in the affected knee joints of transgenic Del1 mice using their non-transgenic littermates as controls.
MATERIALS AND METHODS
Experimental animals
This study was conducted on 120 transgenic Del1 male mice, with 120 of their non-transgenic littermates as controls. Mice heterozygous for the Del1 locus, carrying six copies of an engineered 39 kb Col2a1 transgene containing a deletion of exon 7 and intron 7,25 were mated with non-transgenic animals sharing the same C57bl × DBA background. The litters were genotyped by polymerase chain reaction amplification of tail genomic DNA using two oligonucleotide primers flanking the deletion. The animals were killed at birth and at ages 5, 10, 20, and 35 days, and 2, 3, 4, 6, and 9 months, and their knee joints prepared for RNA analyses (six transgenic mice and six controls each time), and for histological and immunohistological analyses.7 Representative sections from a minimum of three animals at each time point and for each genotype were used for every immunoassay. The study protocol was approved by the institutional committee for animal welfare.
Hybridisation probes
cDNA clones for mouse MMP-13 and MMP-14 mRNAs were constructed using the reverse transcription-polymerase chain reaction (RT-PCR) method and total RNA from mouse cartilage and liver as templates. Random hexamers and oligo(dT) were used to prime reverse transcription of 1 μg of total RNA by Maloney murine leukaemia virus reverse transcriptase under conditions suggested by the supplier (Gibco BRL, Gaithersburg, MD, USA). Aliquots of cDNA were used for amplification by the PCR (AmpliTaq, Perkin Elmer, Branchburg, NJ, USA) using oligonucleotide primers based on existing mouse MMP-13 and MMP-14 sequences27,28 defining fragments of 1424 bp and 584 bp, respectively. The reactions were cycled through denaturation at 94°C for one minute, annealing at 54°C for two minutes, and extension at 72°C for two minutes. After 30 amplification cycles, aliquots of the reactions were fractionated on 1.0% agarose gels, the specific fragments purified and cloned by ligation into the pGEM-T vector (Promega, Madison, WI, USA). The cloned fragments were sequenced using ABI PRISM 377 DNA sequencer. The cDNA clones obtained for mouse MMP-13 and MMP-14 were named pMMMP-13-1 and pMMMP-14-1, respectively.
Northern hybridisation
Total RNA was isolated from knee joints of Del1 and control mice at 10 different ages, as described above. As it was impossible to obtain enough RNA from isolated articular cartilage, whole knee joints containing tibial and femoral epiphyses (dissected at the level of growth plates), the menisci, patella, and ligaments, were used for isolation of total RNA. After dissection the samples were frozen immediately in liquid nitrogen, pulverised, and extracted using the guanidinium isothiocyanate method.29 Aliquots (10 μg) of total RNA were denatured with glyoxal and dimethylsulphoxide, electrophoresed on 0.75 % agarose gels, transferred by blotting onto Pall Biodyne membranes, and hybridised with 32P labelled probes for MMP-2, -3, -8, -9, -13, and -14 mRNAs, using clone pK-191,30 clone 63083,31 a subclone of clone I139A1,32 M92 KD-1,33 pMMMP-13-1 (above), and pMMMP-14-1 (above), respectively. For TIMP-1, -2, and -3 mRNAs, plasmids pMTIMP-1-1, pMTIMP-2-1, and pMTIMP-3-1 were used, respectively.34 After high stringency washes, the bound radioactivity was detected and quantified on a Fuji Bas 5000 phosphoimager (Fuji, Tokyo, Japan) and Tina 2.0 software package (Raytest Isotopen Messgeräte QmbH, Straubenhardt, Germany). Statistical differences in the mRNA levels between genotypes were tested by Student's t test at each time.
Antibodies
Two polyclonal antibodies MV-1 and MV-2 were used to localise type II collagen neoepitopes in articular cartilage.12 These antibodies were raised against synthetic peptides LAGQRGIVGC and QRGIVGLPGC, which correspond to the eight N-terminal amino acids of the one quarter fragment of type II collagen after MMP-1 and MMP-13 cleavage, respectively, with an extra G as a spacer and C for coupling the peptides to carrier. The specificity of these antibodies has been tested earlier in human tissue.12 The distribution of MMP-13 was studied with a polyclonal antibody specific to a synthetic peptide of 26 amino acid of human MMP-13 (BIOTREND Chemikalien, Köln). Distribution of TIMP-1 was studied with a rabbit polyclonal antibody against human TIMP-1 (BIOTREND Chemikalien, Köln).
Immunohistochemistry
Dissected limbs were fixed in 4% paraformaldehyde, demineralised in 10% EDTA for 5–20 days, dehydrated, embedded in paraffin, and sectioned sagitally. Histological sections of the knee joints were either stained with haematoxylin and eosin, or processed for immunohistochemistry. Histological evaluation of the knee joints showed progressive erosion and degradation of articular cartilage, which were graded into five categories as described earlier.7,35 For immunohistochemistry, 5 μm sagittal sections from central regions of condyles were deparaffinised, rehydrated in a series of descending ethanol concentrations, and digested for one hour with testicular hyaluronidase (2 mg/ml) (Sigma, St Louis, MO) in phosphate buffered saline (PBS) (pH 5). Immunohistochemistry for TIMP-1, MV-1, and MV-2 was performed using the avidin-biotin complex method (Histostain-Plus kit, Zymed, South San Fransisco, CA). Appropriate dilutions (1:50 and 1:100) of specific antisera were applied in PBS containing 1% bovine serum albumin, and the sections incubated overnight at 4°C. After rinses with PBS, a biotin conjugated secondary antibody was applied and incubated for 10 minutes at room temperature. The slides were washed twice with PBS, and then incubated with streptavidin conjugated horseradish peroxidase for 10 minutes. Colour was developed with diaminobenzidine, and the sections counterstained with haematoxylin. For control staining, preimmune serum was used instead of the primary antibody.
Immunohistochemistry for MMP-13 was performed using biotin-labelled antisheep secondary antibody and alkaline phosphatase conjugated streptavidin (Links and Label kit from BioGenex, San Ramon, CA), both for 20 minutes at room temperature. Thereafter the sections were incubated for 10 minutes in colour reaction solution containing 0.2 mg/ml naphthol AS-MX, 1 mM levamisole (Sigma), and 0.1 M Tris-HCl (pH.8.2). Immediately before use Fast Red TR (Sigma) was added to a final concentration of 1 mg/ml. For control staining no secondary antibody was used. Finally, the sections were counterstained with haematoxylin.
RESULTS
Expression of MMP and TIMP mRNAs in the knee joints of Del1 and control mice
Expression patterns of mRNAs for MMP-2, -3, -8, -9, -13, and -14 in the knee joints of transgenic Del1 mice and controls were studied by Northern analysis between birth and the age of 9 months (fig 1). The highest levels of MMP-2 mRNA were seen during the period of rapid epiphysial growth, between the ages of 20 days and 3 months (fig 1A). In the older animals, the mRNA levels were below the detection limit. The levels of MMP-3 mRNA peaked at the age of 20 days and thereafter gradually declined (fig 1B). The mRNA expression of MMP-8 was low until one month of age, whereafter it steadily increased with aging (fig 1C). More variation was seen in the mRNA levels for MMP-9 at different ages (fig 1D). After an initial peak at the age of 20 days, the levels declined, but increased again in skeletally mature animals both in Del1 and control mice. During advanced knee joint degeneration at the age of 9 months, MMP-9 mRNA levels of Del1 mice were significantly higher than those of controls (p<0.05). At this age, cartilage erosion in Del1 mice had advanced to subchondral bone with signs of bone remodelling.

Compiled results of Northern analyses of knee joints for MMP-2 (A), MMP-3 (B), MMP-8 (C), MMP-9 (D), MMP-13 (E), MMP-14 (F), TIMP-1 (G), TIMP-2 (H), and TIMP-3 (I) mRNA levels. Total RNAs isolated from knee joints of transgenic Del1 mice and non-transgenic controls at different ages shown below the columns (NB, newborn; d, day; m, month) were analysed by Northern hybridisation. After hybridisation with cDNA probes which detected specific mRNAs of various MMPs and TIMP-1, -2, and -3, the hybridisation intensities were quantified by phosphor imaging and normalised per amount of 28S rRNA determined by hybridisation. The results are shown as relative units. Open columns denote control samples (n=6 at each time/age group), and black columns Del1 samples (n=6 at each time/age group). Statistical significance of the difference in mRNA levels between the control and Del1 mice is shown above the columns (Student's t test; *p<0.05; **p<0.01).
The expression pattern of MMP-13 mRNA (fig 1E) most clearly related to the development of cartilage degeneration, which starts at 3-4 months. A significant increase in MMP-13 mRNA levels was seen between transgenic Del1 and control mice at the onset of articular cartilage degeneration at the age of 3 months (p<0.01). The mRNA levels for MMP-14 remained relatively stable throughout growth and aging both in Del1 mice and in their littermates, although at the age of 5 days, a statistically significant difference was seen between the groups (fig 1F).
Each of the three TIMP mRNAs also exhibited individual expression patterns in the knee joints. The highest levels of TIMP-1 mRNA were seen between 10 days and 4 months of age—for example, during endochondral ossification and growth of the epiphysial heads. During this period, TIMP-1 mRNA levels in Del1 mice were significantly lower than in control mice (fig 1G). TIMP-2 mRNA levels increased throughout the growth period. In the controls, the maximal level was reached at the age of 3 months, and in Del1 mice at the age of 4 months (fig 1H). TIMP-3 mRNA levels were relatively stable at all times, except for an increase at the age of 10 days, with significantly lower values in Del1 mice (fig 1I). Overall, the mRNA levels for the three TIMPs were lower in the knee joints of Del1 mice than in those of control mice.
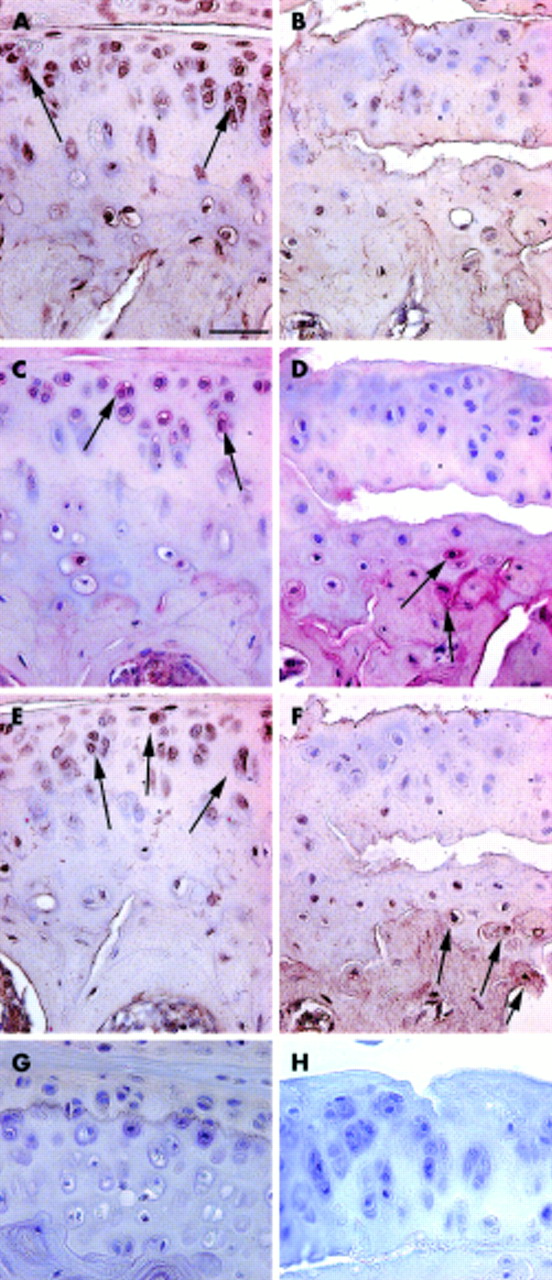
Immunolocalisation of type II collagen neoepitopes, MMP-13 and TIMP-1 in degenerating articular cartilage
In control mice, intracellular and pericellular immunostaining for type II collagen neoepitopes was predominantly seen in chondrocytes in the superficial and middle zones of uncalcified cartilage (fig 2A). MMP-13 and TIMP-1 were localised in the same regions (figs 2C and 2E, respectively). In Del1 mice, the first superficial fibrillations were seen at the age of 3 months. In some specimens, uncalcified cartilage was detached from calcified cartilage in central areas of the condyles (fig 2B). In such areas, no neoepitopes were seen in the uncalcified cartilage (fig 2B). As the distribution patterns seen with MV-1 and MV-2 neoepitope antibodies were essentially identical, only the former is shown. Strong staining for MMP-13 was seen in deep calcified cartilage, and in the adjacent subchondral bone (fig 2D). TIMP-1 immunostaining was seen in subchondral bone matrix (fig 2F) in the same areas where MMP-13 was present.

Immunolocalisation of type II collagen neoepitopes (A, B), MMP-13 (C, D), and TIMP-1 (E, F) during early stages of cartilage degeneration. Tissue sections from 3 month old Del1 (B, D, F) and control (A, C, E) mice. In control mice, type II collagen neoepitopes (A, arrows) and MMP-13 (C, arrows) were detected intracellularly and pericellularly in the uncalcified cartilage. In Del1 mice, MMP-13 was mainly localised in deep calcified cartilage and in subchondral bone (D, arrows). Also, TIMP-1 immunostaining was seen in the matrix of subchondral bone in Del1 mice (F). The distribution of TIMP-1 in control mice (E, arrows) resembled that of the type II collagen neoepitopes and MMP-13. Paraffin sections; avidin-biotin complex with horseradish peroxidase label and DAP detection (A, B, E, F) or alkaline phosphatase label and Fast Red detection (C, D), counterstaining with haematoxylin. Staining controls using normal rabbit serum (G) and without secondary antibody (H). The bar in panel A=25 μm.
By the age of 9 months, severe degeneration of articular cartilage was seen in all Del1 mice. The progression of degeneration was slower in control mice, and corresponded approximately to the level seen in Del1 mice at the age of 3 months. At 9 months, only a weak staining for neoepitopes and MMP-13 was seen in chondrocytes of Del1 mice located in severely eroded cartilage. In areas more distal to the erosions, neoepitopes were detected both intracellularly and pericellularly in uncalcified articular cartilage (data not shown). More intense staining was seen in deep calcified cartilage and adjacent subchondral bone (data not shown).
Hyperplasia of synovial membrane and immunostaining with MMP-13 and TIMP-1 antibodies
Synovial proliferation at the anterior margin of lateral tibia and femur was evaluated by counting the number of cell layers in 4 month old mice. In control mice, the synovial membrane was one to two cell layers thick (figs 3A and C), whereas in Del1 mice it contained three to four layers (figs 3B and D). In addition to increased thickness, the synovial membrane of Del1 mice exhibited stronger staining for MMP-13 (fig 3B) and TIMP-1 (fig 3D). This difference persisted at the age of 9 months (not shown).
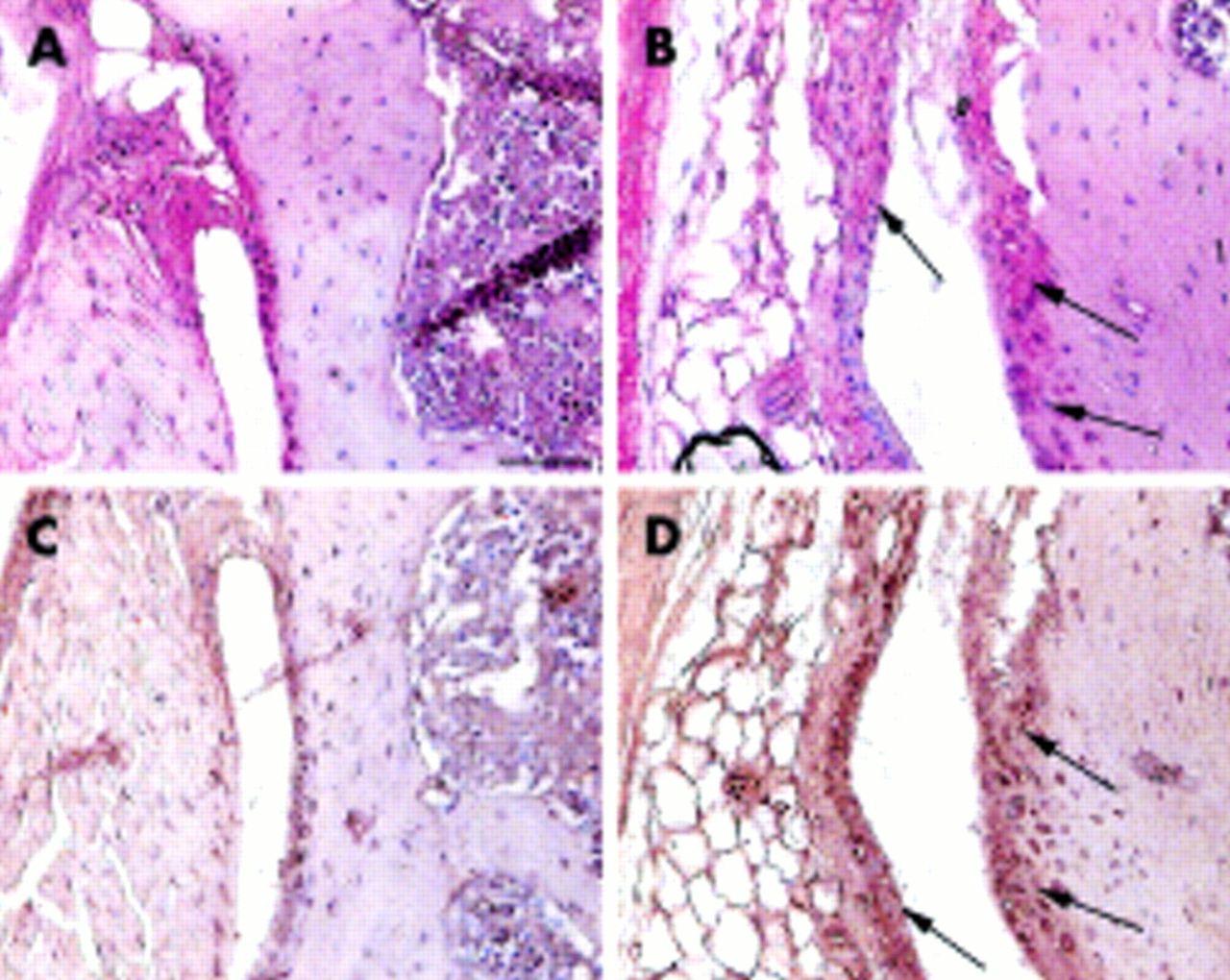
{kind=link}
{kind=link}
{kind=link}
Appearance of the synovial membrane in control (A, C) and Del1 mice (B, D) at the age of 4 months. In Del1 mice, the synovial membrane of the knee joint was thickened and the immunostaining for MMP-13 (B, arrows) and TIMP-1 was increased (D, arrows) compared with the control knee joint (A, C, respectively). Paraffin sections; avidin-biotin complex with horseradish peroxidase label and DAP detection (C, D) or alkaline phosphatase label and Fast Red detection (A, B), counterstaining with haematoxylin. The bar in panel A=50 μm.
DISCUSSION
Several of the MMPs analysed in this study have been implicated directly or indirectly in the degradation of fibrillar collagens. All three collagenases, MMP-1, MMP-8, and MMP-13, have been shown to cleave type II collagen into specific 1/4 and 3/4 fragments, resulting in the formation of characteristic neoepitopes that can be detected by specific antibodies.11,12 Although the existence of a true murine orthologue of the MMP-1 gene remains questionable,36 the initial cleavage of collagen fibrils and generation of neoepitopes in mouse cartilage can definitely be achieved by the other interstitial collagenases, MMP-8 and MMP-13, and possibly by MMP-14. Although it has been shown that MMP-8, a neutrophil collagenase, is also produced by chondrocytes,37,38 most data on human OA point to the presence of increased MMP-13 levels as important in development of the disease.39–41 MMP-13 also has higher catalytic activity towards type II collagen than towards other fibrillar collagens, whereas MMP-8 preferentially cleaves type I collagen.42,43
In the present model, MMP-13 was the only MMP showing a significant OA related change in its mRNA expression pattern between Del1 and control mice. Despite the temporal association of increased MMP-13 expression and onset of OA degeneration in the knee joints of Del1 mice, several lines of evidence suggest that MMP-13 does not participate directly in the initiation of cartilage degeneration. Thus, no immunostaining for MMP-13 was seen at sites of superficial fibrillation. Neither were type II collagen derived neoepitopes concentrated in these areas. Overall, the faint staining intensity for type II collagen neoepitopes in articular cartilage is in accordance with the very slow turnover rate of type II collagen in adult cartilage.44 The increased staining for MMP-13 in Del1 knee joints was localised in the hyperplastic synovial membrane, in subchondral bone, and in deep layers of articular cartilage. We interpret the up regulation of MMP-13 expression in synovial tissue as representing a secondary inflammatory response to degradation products released from articular cartilage into the joint cavity. Substantial up regulation of MMP-13 mRNA and activation of the enzyme has been seen in synovial tissue in response to inflammatory mediators.45 Thickening of the synovial membrane with associated expression of MMP-13 by synovial fibroblasts has also been found in human OA.15,46 The finding that subchondral bone and deep layers of articular cartilage are other important sources of MMP-13 during cartilage degeneration is interesting as this may explain the subchondral bone remodelling often seen both in human OA47,48 as well as in the present model.7
Considering that the primary defect in transgenic Del1 mice is a type II collagen mutation, it is not surprising that no evidence for MMP mediated degradation was found in the early cartilage fibrillations in Del1 mice. Cartilage degeneration in these mice is obviously related to reduced structural integrity of hyaline cartilage, which must make the articular cartilage less resistant to physical trauma. The mechanism whereby structural weakening of articular cartilage activates MMP-13 production and initiates matrix degradation at the cartilage-bone interphase, remains unknown.
The intracellular and pericellular localisation of both the neoepitopes and MMP-13 in uncalcified cartilage of Del1 and control mice was surprising. We believe that the lack of immunostaining in cartilage matrix reflects the very slow turnover rate of type II collagen in this tissue.44 In Del1 mice the cell associated immunostaining for MMP-13 and type II collagen neoepitopes probably reflects rapid processing of the mutant collagen produced. Similar distribution of epitopes in control mice suggested that this largely represents normal post-translational quality control.
Although several different expression patterns were found for the other MMP mRNAs studie, none of them showed changes that could be related to articular cartilage degeneration. The mRNA levels of MMP-8 steadily increased upon aging but were not dependent on OA. The expression of MMP-2 and MMP-3 mRNAs peaked during the period of endochondral growth, whereas MMP-9 mRNA levels increased as the joint degenerated. At the age of 9 months, MMP-9 mRNA levels were significantly higher in Del1 mice than in controls. By this time, cartilage erosions in Del1 mice had advanced to subchondral bone, and the increase in MMP-9 production was probably a sign of bone remodelling. In the present model, the enzyme was predominantly located on endosteal bone surfaces, and was rarely seen in articular chondrocytes (data not shown). In human OA, however, increased MMP-9 mRNA levels have been seen in articular cartilage chondrocytes.49,50 The mRNA levels for MMP-14 remained relatively stable through growth and aging, with no significant OA related changes between Del1 mice and their littermates. The expression profiles of MMP mRNAs thus suggest that these enzymes do not play a major part in the initial pathogenesis of cartilage degeneration of Del1 mice. Conflicting findings on MMP levels have also been made in human OA and in experimental animal models for OA.39,49,51
We earlier reported the differential expression patterns of TIMP mRNAs during normal growth and aging of the skeleton.34 In this study, TIMP-1 mRNA levels were consistently lower in the knee joints of Del1 mice than in those of controls. Immunohistochemistry confirmed that the inhibitor was mainly derived from synovial tissue. On the contrary, increased levels of TIMP-1 mRNA have been found in human OA.52 In this study, the mRNA levels for TIMP-2 and TIMP-3 were generally lower in Del1 mice than in their non-transgenic littermates. It has been suggested that an imbalance between MMP-13 and its inhibitors TIMP-1 and TIMP-2 contributes to progression of knee joint degeneration in human OA joints.15,22,52 Although correction of the imbalance between MMPs and TIMPs is an attractive new concept to treat human OA, the results obtained in the present study suggest that Del1 mice are of limited value for “proof of concept” testing of such treatments, as MMPs do not appear to have a central role in the initiation of articular cartilage degeneration in this model. However, they may have potential importance in the progression of the defects.
Acknowledgments
The expert technical help of Merja Lakkisto and Tuula Oivanen is gratefully acknowledged. This study was supported by the Academy of Finland (project no 52940). We thank Carlos Lopez-Otin for kindly providing us with the plasmid for mouse MMP-8. Heli Salminen has been a recipient of a training grant from the Turku Graduate School of Biomedical Sciences.