Article Text

Abstract
OBJECTIVE Molecular biology techniques such as polymerase chain reaction (PCR) and ligase chain reaction (LCR) are routinely used in research for detection ofC trachomatis DNA in synovial samples, and these methods are now in use in some clinical laboratories. This study aimed at determining the method best suited to molecular diagnosis ofC trachomatis by examining four standard DNA preparation methods using chlamydia spiked synovial tissue and chlamydia infected monocytes.
METHODS Synovial tissue from a chlamydia negative patient with rheumatoid arthritis was spiked with defined numbers of C trachomatiselementary bodies (EB). Purified human peripheral monocytes from normal donors were infected with the organism at a multiplicity of infection 1:1 in vitro and harvested after four days. DNA was prepared from all samples by four methods: (1) QIAmp tissue kit; (2) homogenisation in 65°C phenol; (3) incubation at 97°C; (4) proteinase K digestion at 97°C. DNA from methods 1 and 2 was subjected to PCR using two different primer sets, each targeting the C trachomatis omp1 gene. LCR was done on DNA prepared by each method.
RESULTS In synovial tissue samples spiked with EB, and in monocytes persistently infected with the organism, preparation of template using the QIAmp tissue kit (method 1) and the hot phenol extraction technique (method 2) allowed sensitive detection of C trachomatis DNA. These methods also produced template from both sample types for LCR. DNA prepared by heat denaturation (method 3) allowed only low sensitivity chlamydia detection in LCR and did not work at all for PCR. Proteinase K digestion plus heat denaturation (method 4) gave template that did not allow amplification in either PCR or LCR assays.
CONCLUSIONS The sensitivity of detection for C trachomatisDNA in synovial tissue by PCR and LCR depends strongly on the method used for preparation of the amplification template. LCR targeting the multicopy chlamydial plasmid and two nested PCR assay systems targeting the single copy omp1 gene showed roughly equivalent sensitivity. Importantly, template preparation method and the specific PCR primer system used for screening must be optimised in relation to one another for highest sensitivity.
- polymerase chain reaction
- ligase chain reaction
- DNA preparation
- C trachomatis
Statistics from Altmetric.com
The intracellular bacterial parasiteChlamydia trachomatis has long been associated with the pathogenic process ending in reactive arthritis (ReA) and Reiter's syndrome (RS; reviewed by Köhleret al 1). Standard laboratory culture of synovial tissue for the organism, however, as well as direct fluorescent antibody screening (DFA), are often negative.2 ,3 Our observations indicate that synovial culture negativity for chlamydia results primarily from the paucity of new infectious elementary bodies (EB) produced by the organism during persistent synovial infection.4 ,5 We also showed that DFA negativity of chlamydia infected synovium results from the selective downregulation of expression of the omp1gene encoding the chlamydial major outer membrane protein during persistent infection6; this protein is the target of most monoclonal antibodies (mAbs) used in DFA.
Regardless of the unusual molecular genetic characteristics adopted by persistent synovial C trachomatis, it is clear from our observations and those of others that the number of bacterial cells present in the inflamed joints of patients with ReA/RS can be extremely low.7 For this reason, and because of the culture negativity and DFA negativity of most samples, polymerase chain reaction (PCR), and, more recently, ligase chain reaction (LCR), screening systems have become the preferred methods used in research laboratories to assess the presence of chlamydia in synovial materials. For PCR, some commercial systems are available for clinical laboratory use in chlamydia detection (for example, Amplicor), though at least some of these have been shown to be less sensitive than PCR assays designed for research use (APH, unpublished observations). At present, however, no standardised system for chlamydia directed PCR screening has been adopted by groups participating in arthritis studies; that is, each laboratory prepares DNA from clinical or other samples by its own method, and primer systems and the chlamydia DNA sequences targeted, vary widely among research groups. This is also true for LCR analysis targeting chlamydial DNA, though commercial LCR systems do exist and are in use.
Any brief survey of the literature relating to ReA/RS indicates that the percentage of patient samples that are PCR positive (or LCR positive) for C trachomatis DNA varies among laboratories.8 ,9 To some extent this variation is due to the fact that some laboratories analyse DNA from synovial fluid (SF), whereas others use template prepared from synovial tissue. We previously examined which DNA preparation method provides the best template for PCR analyses of DNA from SF samples10 (see also “Discussion”), and our results were consistent with those of other groups that noted the relevance of optimised template preparation techniques for analysis of SF.11-13 However, Braniganet al have reported that synovial tissue allows higher sensitivity than SF in detection of C trachomatis.8 We therefore analysed synovial tissue using both samples spiked with C trachomatisEB, and persistently infected human monocytes, to define the optimal template preparation method(s) for molecular analyses. We also tested two different but commonly used PCR assays, and a commercial LCR assay, to define which template preparation method provides the best amplification substrate for each assay type. The results presented here indicate that it is critically important to optimise the DNA preparation method and the specific primer system together to achieve the highest sensitivity in both PCR and LCR.
Methods
SYNOVIAL TISSUE SAMPLES
Multiple synovial tissue samples were procured from a patient with rheumatoid arthritis (RA) by a standard biopsy procedure using a Parker-Pearson needle.14 DNA prepared from a portion of this sample was analysed by PCR before addition of EB (see below), to ensure that it was PCR negative for chlamydia DNA.
PREPARATION OF CHLAMYDIA
C trachomatis EB (serovar K) were cultured in Hep-2 cells as described.15 Serovar K was chosen because it causes urogenital infection and has been shown to induce ReA. EB were purified in a discontinuous gradient of Urografin (Schering, Berlin, Germany) by ultracentrifugation, as described.15 EB so prepared were resuspended in 1 ml sucrose phosphate buffer (0.01 M sodium phosphate, 0.25 M sucrose, 5 Ml-glutamic acid pH 7.2; Sigma, St Louis, USA) and stored at −80°C. Each preparation was analysed by titration on Hep-2 cells and subsequent indirect immunoperoxidase assay, then adjusted to 2 × 107 infection forming units (IFU)/ml. IFU represent the number of infective chlamydia in a given sample. The EB stock was diluted 100-fold, aliquoted, and stored at −80°C. For each assay, one aliquot was thawed and further diluted in 0.9% NaCl, 0.5 mg/ml bovine serum albumin (BSA) for use.
ADDITION OF ELEMENTARY BODIES TO SYNOVIAL TISSUE BEFORE ANALYSIS
Spiking of synovial tissue samples with known numbers of EB was done as described.6 ,8 Briefly, aliquots of pure EB were thawed and diluted to 20, 30, 40, 60, 80 IFU/10 μl. Three slides were made from each dilution, and each was analysed by immunofluorescence to determine the number of chlamydia EB/IFU in each dilution; the murine mAb used in these determinations was from the Micro-Trak system (Syva Corp, Palo Alto, CA, USA). Samples were analysed using an epifluorescence microscope (Leitz, Wetzlar, Germany). On average, 13 particles corresponded to 1 IFU in each dilution (slope = 13;r 2 = 0.45; p=0.0001). EB in known numbers were added to synovial tissue samples (5 mg/sample) in a 10-fold decreasing series ranging from 105 to 10−2 per synovial sample. One tissue sample containing no added EB was processed in each experiment as a negative control. After addition of EB to tissue samples, each preparation was centrifuged at 60 000 × g for 30 minutes at 4°C, and the resulting pellet was processed by the different methods for total DNA preparation given below.
PERSISTENT INFECTION OF MONOCYTES; SERIAL DILUTIONS
For some experiments, human peripheral monocytes were prepared from volunteer blood samples by the standard method, as described.16 ,17 These monocytes were infected withC trachomatis K serovar at a multiplicity of infection 1:1 (that is, 1 chlamydia EB/monocyte). The relative number of chlamydia infected monocytes in each preparation was determined as above for the EB. Briefly, aliquots of infected cells were analysed by immunofluorescence to determine the number of infected monocytes in each preparation; the murine mAb used in these determinations again was from the Micro-Trak system (Syva Corp, Palo Alto, CA, USA). Samples were analysed with an epifluorescence microscope. On average, approximately 1% of monocytes was infected in each preparation analysed. At four days after infection the cells were harvested, serially diluted in 10-fold steps in 0.9% NaCl + 0.5 mg/ml BSA, then mixed with synovial biopsy tissue. Infected monocytes were processed by each of the four DNA preparation methods used in the EB studies.
DNA PREPARATION METHODS
Total DNA was prepared from each spiked synovial tissue sample and from chlamydia infected human monocytes by each of four different methods, then resuspended in 50 μl of TE buffer (10 mM Tris-HCl pH 8, 1 mM EDTA); 5 μl of each DNA preparation was used for each PCR or LCR analysis.
Method 1 used the commercial DNA purification kit supplied by Qiagen (Qiagen GmbH, Hilden, Germany) and designated the QIAmp tissue kit; preparations were done according to the manufacturer's instructions. Briefly, the synovial tissue/EB pellets were incubated in the supplied lysis buffer containing proteinase K at 55°C overnight. DNA was isolated over the column supplied, then eluted, and precipitated overnight at −70°C in three volumes 100% ethanol; DNA so prepared was resuspended in 50 μl TE buffer.
Method 2 was the standard method of preparation by tissue homogenisation in hot buffered phenol.6 ,8 ,18 Briefly, samples were minced while still frozen and promptly homogenised in a Dounce containing 1 ml phenol plus 0.5 ml of aqueous buffer (25 mM Tris-HCl pH 8; 10 mM EDTA, 100 mM NaCl, 0.5% sodium dodecyl sulphate) preheated to 65°C. After homogenisation, mixtures were centrifuged, and the aqueous phase containing nucleic acids was collected, then extracted twice with chloroform:isoamyl alcohol (24:1). Total DNA was obtained by precipitation in ethanol and resuspension in 50 μl TE buffer.
Method 3 simply involved sample incubation at 97°C for 15 minutes in a water bath, followed by resuspension in LCR reaction buffer (see below).
Method 4 consisted of sample digestion with proteinase K (concentration not given in the kit) in lysis buffer supplied in the QIAmp tissue kit overnight, followed by incubation at 97°C for 15 minutes and resuspension in LCR reaction buffer. Five independent repeats of each of the four extraction methods were done, using independent serial dilutions of EB and infected monocytes in each case.
PCR AND LCR ANALYSES
Template DNA from both synovial tissue/EB mixtures and chlamydia infected human monocytes as prepared by methods 1 and 2 was subjected to PCR analyses using two different, independently developed primer sets. The first primer system (set 1) targets the C trachomatis major outer membrane protein gene (omp1), as described by Boboet al 19; all assays were done using conditions described by Kuipers et al.20 The second primer system (set 2) targets a different DNA sequence within the C trachomatis omp1 gene and was developed in one of our laboratories (APH); that system, and the conditions for its use, have been described.6 ,8 ,18Primers used in both PCR assays were synthesised by BRL (Gaithersburg, MD, USA). All amplifications were done in an MJ PT-100 thermocycler (Watertown, MA, USA). The LCR system used for the present analyses was the standard commercial kit supplied by Abbot Laboratories (Abbot Park, IL, USA). The oligonucleotides used by this kit target the 7 kbp plasmid of C trachomatis; LCR reactions were done as specified by the manufacturer.
STATISTICAL ANALYSIS
Definition of the number of EB relative to IFU was done by standard regression analysis. For PCR and LCR assays, determination of the statistical significance of sensitivity relations for the different extraction methods using the two primer sets was made by the Kruskal-Wallis test, followed by the Mann-Whitney U test. A value of p⩽0.05 was considered significant in all such analyses. EB measured by immunofluorescence were the basis for determining sensitivity. Because some EB may become disrupted during isolation, the amount of chlamydia DNA present in each sample may be somewhat higher than given here. However, because all analyses were performed with the same stock of EB, comparative analysis is justified. For comparison, sensitivity is given for each method as the number of EB detected per sample.
Results
SENSITIVITY OF CHLAMYDIA DIRECTED PCR SCREENING FOR ELEMENTARY BODY DNA AS A FUNCTION OF TEMPLATE PREPARATION METHOD
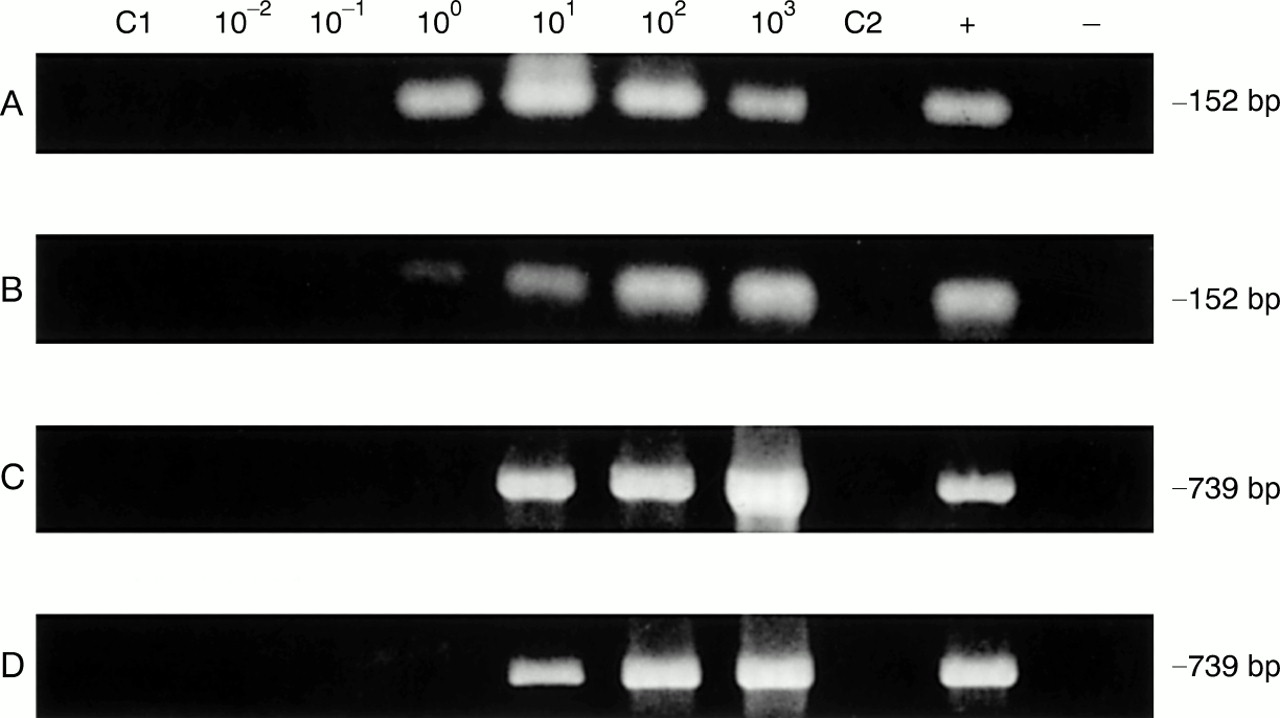
The control PCR analyses displayed in fig 1 (lanes C1 and C2 in each panel) show that the synovial tissue sample used in the spiking experiments given below was negative for C trachomatis omp1 DNA with each of the two primers systems. Using template DNA prepared by the QIAmp tissue kit (method 1) and amplified by PCR primer set 1, we repeatedly found as few as 10 EB/sample in assays of five independent serial dilutions (table 1; fig 1A). With DNA prepared through hot phenol (method 2) and amplified with the same primers, sensitivity was approximately 100 EB/sample analysed (four assays 100 EB/sample, one assay 10 EB/sample; fig 1B); this difference was not significant (p=0.589). Interestingly, the DNA prepared by method 1 and amplified using set 2 primers showed a lower sensitivity (100 EB/sample; four assays 100 EB/ sample, one assay 10 EB/ sample) than the same DNA amplified with set 1 primers (fig 1C; not significant, p=0.945). Conversely, DNA prepared by method 2 and amplified with primer set 2 was routinely able to detect a single EB/sample in each of five repetitions of the experiment (fig 1D). Again, however, the difference was not significant (p=0.620); comparison of the sensitivity of the template prepared through hot phenol with set 1 v set 2 primers also showed no significant difference (p=0.366). DNA prepared by either simple incubation of spiked tissue samples at 97°C (method 3) or extensive proteinase K digestion combined with high temperature incubation (method 4) were not assayable by PCR because of the buffer constituents in the preparative mixture. Thus PCR template DNA prepared by the QIAmp tissue kit gave slightly higher sensitivity with set 1 primers than with set 2 primers, and template prepared by the hot phenol method showed the reverse. However, both methods produced high quality DNA for use as template in PCR amplifications.

Representative polymerase chain reaction sensitivities in detection of C trachomatis in synovial tissue samples spiked with elementary bodies (EB) using template DNA prepared by the QIAmp tissue kit (method 1) and homogenisation in hot phenol (method 2), with set 1 and set 2 primers. (A) DNA preparation method 1 with primer set 1; (B) DNA preparation method 2 with primer set 1; (C) DNA preparation method 1 with primer set 2; (D) DNA preparation method 2 with primer set 2. Assays were performed as given in “Methods”; amplification product size in base pairs (bp) is given at the right. In each panel lanes C1 and C2 used template DNA from synovial tissue of the patient with rheumatoid arthritis without added EB, and primer sets 1 and 2, respectively. The positive (+) control and negative (−) control lanes contained pure EB DNA and water only, respectively, as amplification template.
Sensitivity of polymerase chain reaction for the detection of C trachomatis in synovial tissue depending on the sample preparation and primer set used. Synovial tissue was spiked either with isolated elementary bodies (EB) or with C trachomatis persistently infected monocytes
SENSITIVITY OF CHLAMYDIA DIRECTED LCR SCREENING FOR ELEMENTARY BODY DNA AS A FUNCTION OF TEMPLATE PREPARATION METHOD
The LCR assay used in these studies is a commercial kit currently in use in some clinical laboratories. Template DNA prepared by method 1 gave a routine sensitivity of approximately 100 EB/sample (three assays 100 EB/ sample, one assay 10 EB/ sample, one assay 1 EB/sample), whereas using template prepared by method 2 we were able to detect about 10 EB with this assay system (table 2; p>0.05). Treatment of the spiked tissue samples with high temperature incubation alone (method 3) produced template that gave a sensitivity of about 100 EB/sample in each of five preparations analysed; this difference was not significant when compared with LCR results using DNA prepared by method 1 (p>0.05), but it was significant when compared with results using DNA prepared through hot phenol (p<0.01). Because the digestion buffers used for proteinase K treatment in method 4 included constituents that precluded LCR amplification, template prepared by this method gave no result. Thus as with the PCR analyses above, the sensitivity of molecular diagnosis of chlamydia by LCR is influenced by the means used to prepare amplification template from clinical samples.
Sensitivity of ligase chain reaction for the detection of C trachomatis in synovial tissue depending on the sample preparation. Synovial tissue was spiked either with isolated elementary bodies (EB) or with C trachomatis persistently infected monocytes
SENSITIVITY OF CHLAMYDIA DIRECTED PCR AND LCR SCREENING FOR RETICULATE BODY DNA AS A FUNCTION OF TEMPLATE PREPARATION METHOD
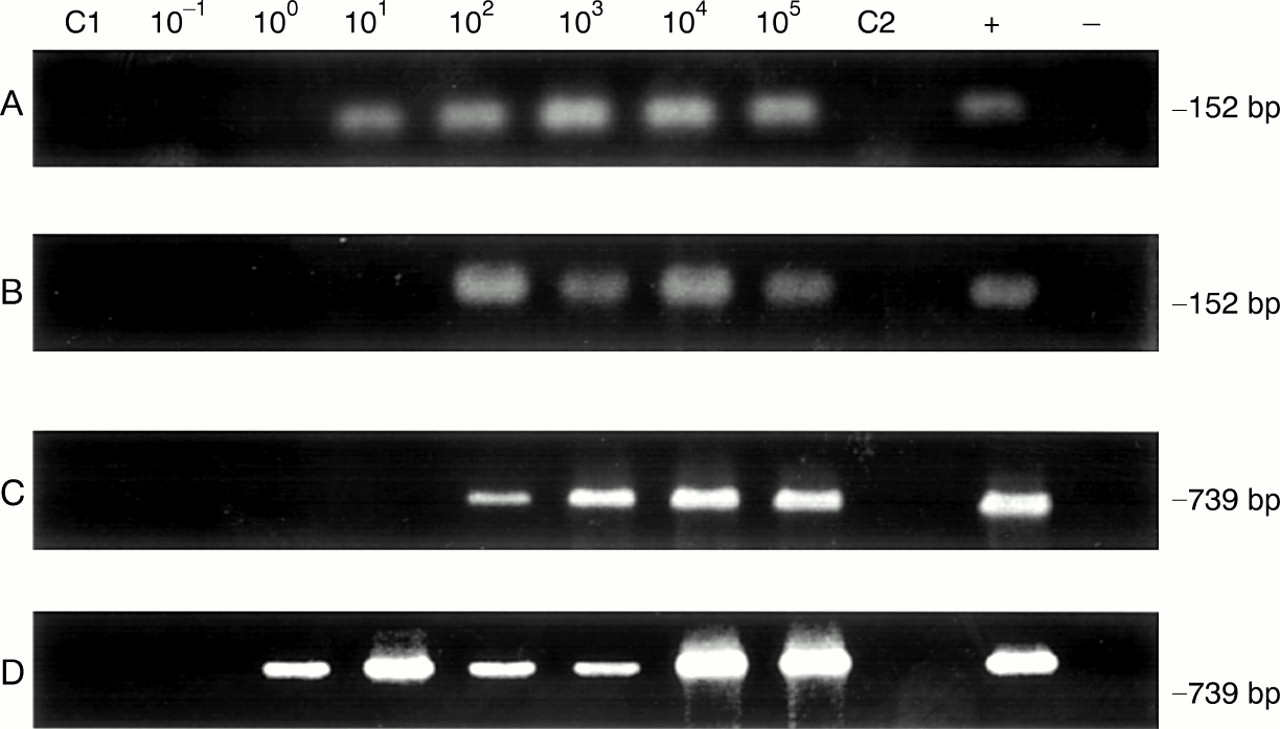
Chlamydia EB are the extracellular form of the organism, and they possess an extremely durable cell wall. During synovial infection the organism is present in joint tissue, primarily in the intracellular, reticulate body-like form, which lacks a substantial cell wall. To determine whether the methods used for DNA preparation from EB are equally effective in template preparation from intracellular chlamydia, serially diluted, cultured human peripheral monocytes persistently infected with C trachomatis were mixed with synovial biopsy samples. These were processed by each of the four DNA preparation methods above; as in the EB directed studies, template DNA from methods 1 and 2 were subjected to PCR using primer sets 1 and 2; template prepared by each of the four methods was subjected to LCR also. Using the QIAmp tissue kit and set 1 PCR primers, we detected chlamydia DNA from one infected monocyte (table 1; fig 2A), whereas set 2 primers only identified 10 infected host cells (fig 2C); this was not significantly different. Template DNA from hot phenol treatment gave a sensitivity of one infected monocyte with set 1 PCR primers (fig 2B), and 10 infected cells with set 2 primers (fig 2D), again not significantly different.
{kind=link}
{kind=link}
Representative polymerase chain reaction (PCR) sensitivities in detection of C trachomatis using chlamydia infected monocytes in synovial tissue samples and template DNA prepared by the QIAmp tissue kit (method 1) as well as homogenisation in hot phenol (method 2), with set 1 and set 2 primers. (A) DNA preparation method 1 with primer set 1; (B) DNA preparation method 2 with primer set 1; (C) DNA preparation method 1 with primer set 2; (D) DNA preparation method 2 with primer set 2. Assays were performed as given in “Methods”; amplification product size in base pairs (bp) is given at right. In each panel lanes C1 and C2 PCR was done using the set 1 and set 2 primers, respectively, and template DNA from synovial tissue of the patient with rheumatoid arthritis without added EB. The positive (+) control and negative (−) control lanes contained pure EB DNA and water only, respectively, as amplification template.
LCR analyses showed a sensitivity of one infected monocyte in analyses using template DNA prepared by either method 1 or 2 (table 2). Template prepared by method 3 gave an LCR detection threshold of 10 infected cells; template from method 4 preparation gave no product in LCR assays.
Discussion
Use of chlamydia targeted screening DNA amplification assays is common in research laboratories studying ReA/RS, among other diseases, and use of commercially available amplification assays targeting this and other organisms is becoming more common in clinical laboratories. The commercial assay systems, both PCR and LCR based, include detailed instructions and materials for standardised template preparation, amplification, and product analysis, and thus these systems normally provide reasonably reliable and repeatable screening results. The systems are, however, usually developed for use in detection of chlamydia in anatomical contexts other than synovial materials, such as urine. Our own experience with two of the commercial chlamydia directed PCR systems indicates that although they are quite specific in their targeting, they lack the sensitivity routinely obtained in research derived assays (APH, unpublished observations). For the most part, however, the sensitivity of assays used by researchers has not been standardised, and most target different chromosomal and/or plasmid DNA sequences from the organism. Moreover, virtually every laboratory employs a different method for preparation of template DNA for the amplification process. This diversity of assay and preparative systems has led to diversity in results for the overall percentage of patients with ReA/RS positive for chlamydia DNA in synovium. We recently showed that DNA sample preparation method is crucial in PCR analysis of SF, underlining the need for optimisation and standardisation.10 Other groups have also noted that optimised sample preparation and the choice of the molecular technique greatly influence the sensitivity of detection of C trachomatis in joint material. For example, Wilkinsonet al developed optimised DNA extraction methods and PCR before analysis of SF samples,13 and Baset al demonstrated the effect on assay sensitivity of different template preparation techniques.12
Recently, PCR based analysis of synovial tissue forC trachomatis was shown to give higher sensitivity than a similar analysis of SF.8 To define the importance of the template preparation method in the success/sensitivity of amplification assays, we prepared DNA from synovial tissue/EB mixtures by four standard methods: (1) the QIAmp tissue kit, (2) hot phenol/homogenisation, (3) incubation of tissue/EB mixtures at high temperature, and (4) high temperature incubation combined with proteinase K digestion. We tested DNA prepared by each method in two independently developed PCR systems targeting the same chlamydial gene, and in a commercial LCR system.
The results of our study show that the template preparation method does influence the overall sensitivity of the ensuing amplification process, and in some cases determines whether any amplification product at all is obtained. DNA prepared by the QIAmp kit or by homogenisation through hot phenol yielded template that gave a reasonably sensitive, though not absolutely equivalent, detection of target sequences in both PCR and LCR assays, using DNA from both tissue/EB mixtures and chlamydia infected monocytes mixed with synovial tissue as starting material. PCR and LCR assays of DNA from infected monocytes gave somewhat higher sensitivity than the identical assays of tissue/EB mixtures, of course, because reticulate body-like forms undergoing active, intracellular vegetative growth lack the strong cell wall characteristic of EB. In contrast, high temperature incubation, the method recommended for preparation of urine before PCR, either alone or with proteinase K digestion, of tissue/EB mixtures or C trachomatis infected monocytes produced template that was unusable in PCR, primarily because of the buffer components required. For LCR, high temperature incubation alone produced template that allowed reasonable sensitivity in repeated assays, but similar incubation combined with proteinase K digestion gave template that yielded no LCR product at all from any starting material. Thus although these methods are mechanically simple to perform, they are probably not the methods of choice for template production for either PCR or LCR.
Commercial nucleic acid preparation kits are used by many laboratories for template preparation, and the Qiagen product is among the most commonly used among those. Our results confirm that DNA prepared by this method works well for both PCR and LCR, as expected. The hot phenol method for DNA preparation was among the first described in the literature, and it is still the preferred template preparation method in one of our laboratories (APH). This method, although somewhat laborious and time consuming, is absolutely reliable, produces template of high quality, and generally gives somewhat higher yields than any of several commercial DNA preparation kits (data not shown). In our study template prepared through hot phenol worked about as well overall for both PCR and LCR as that made through the Qiagen product. Thus there seems to be no major advantage to DNA template preparation by either the QIAmp tissue kit or homogenisation in hot phenol for the PCR analyses used in this study (but see below). Given its simplicity, reliability, and non-requirement for toxic chemicals, the Qiagen kit should probably be considered the best preparation method for routine diagnostic purposes, and the hot phenol method the best technique for template to be used for research purposes. However, in other experiments currently in progress we have compared RNA prepared by a Qiagen kit with that derived from DNaseI digestion of total nucleic acids from hot phenol homogenisation of chlamydia infected monocytes, in reverse transcriptase PCR analyses targeting variousC trachomatis transcripts. The results of these initial experiments seem to indicate that RNA prepared by the latter method gave more reliable cDNA preparations, and therefore more sensitive detection, in these assays. Thus whereas DNA prepared by the commercial kit and through hot phenol are more or less equivalent for PCR analysis, the RNA given by the older, more laborious method may be better than that given by the commercial product. We are now in the process of defining precisely how different the quality and recovery of RNA is from these two approaches. Nevertheless, consistent with our previous observations for SF, hot phenol/homogenisation and the commercial kit performed best for DNA extraction.10However, compartment-specific modifications (that is, for SF and synovial tissue) of these two methods are needed for highest sensitivity. Efforts are currently being made in our laboratories to develop an optimised overall method for template preparation and analysis which will perform well for clinical samples from both SF and tissue.
The data shown in tables 1 and 2 indicate that PCR and LCR analyses targeting C trachomatis DNA sequences have roughly the same sensitivity, given constant template input into the assays. This must be understood in terms of the specific target DNA sequences specified for each screening system—that is, the single copyomp1 gene in both nested PCR assays studied here v the multicopy chlamydial plasmid for the LCR system. An LCR system targeting the plasmid probably should show a somewhat higher sensitivity in chlamydia detection than a non-nested PCR based assay targeting a single copy gene sequence, because the plasmid is present in a copy number 10- to 20-fold higher than that of single copy genes in each chlamydial cell. It is because both PCR systems examined here were nested that the sensitivity of the commercial LCR system roughly matched that of the twoomp1 directed PCR systems. However, the template preparation method proved to be an important factor in the sensitivity of chlamydia detection by LCR, as it did for PCR, as discussed next.
We were surprised to find that the detailed sensitivity of the two different PCR assay systems varied somewhat with template preparation method, at least as defined in our EB spiking and infected monocyte experiments. The reason(s) for this difference in sensitivity is not at all clear to us, but it cannot be related to the source of PCR primers (that is, the company synthesising those primers), because both primer sets were prepared by the same source at the same time. Nor can the difference be ascribed to the use of different thermocyclers, because again all amplification reactions were done in the same machine, or to the buffers, DNA polymerase, or other components used in the amplifications themselves. Regardless of the precise reason(s) for the observed differences in sensitivity, however, the results presented here indicate that to achieve maximum sensitivity in PCR screening assays, some care must be taken to optimise use of the chosen primer set in relation to the template preparation method. Although we did not test other commonly used C trachomatisdirected systems or other primer systems, or any of the several other commercial kits for template preparation from tissue or other sources, we suspect that this dictum will be true for them as well as for the systems examined here.
Acknowledgments
The authors gratefully acknowledge the advice of Dr Elke Raum, Department of Epidemiology, Hannover Medical School, in the statistical analyses.
References
Footnotes
This work was supported by grants BMBF No 01 VM 9305–04 (LK), BMBF No 01 VM 9305–05, and 01 GI 9950 (JGK), and NIH grant AR-42541 (APH).