Article Text

Abstract
OBJECTIVE Peripheral T cells from patients with rheumatoid arthritis (RA) are hyporesponsive when stimulated with antigen or mitogen in vitro, possibly owing to increased production of proinflammatory cytokines such as tumour necrosis factor α (TNFα). This study sought to find out if and how RA T cell reactivity is affected during treatment with etanercept (Enbrel), a soluble TNFα receptor.
METHODS Heparinised blood was collected from patients with RA at baseline, after four and eight weeks of etanercept treatment, and from healthy controls. After density separation spontaneous production of interferon γ (IFNγ), TNFα, interleukin 6 (IL6), and IL10 by peripheral blood mononuclear cells (PBMC) was detected by ELISPOT. For detection of T cell reactivity, PBMC were stimulated in vitro with mitogen (phytohaemagglutinin (PHA)), microbial antigens (purified protein derivative (PPD), influenza), or an autoantigen, collagen type II (CII). Supernatants were analysed for IFNγ and IL2 content by enzyme linked immunosorbent assay (ELISA).
RESULTS In RA the number of cells spontaneously producing IFNγ was significantly increased after four, but not eight weeks' treatment with etanercept. T cell reactivity, as measured by IFNγ production to PPD, influenza, and CII was significantly increased after four and sustained after eight weeks' treatment, whereas IFNγ production induced by PHA remained unchanged. TNFα production was significantly higher in patients with RA than in controls and did not change during etanercept treatment.
CONCLUSION Treatment of patients with RA with etanercept may lead to increased peripheral T cell reactivity both to microbial antigens and to self antigens such as CII. These findings indicate that TNFα blockade may not only suppress but also stimulate certain aspects of antimicrobial immune defence and autoimmunity.
- etanercept
- T cell reactivity
- rheumatoid arthritis
- autoimmunity
Statistics from Altmetric.com
Treatment with agents blocking the function of tumour necrosis factor α (TNFα) has proved to be highly effective in ameliorating the symptoms of chronic joint inflammation characteristic of rheumatoid arthritis (RA).1 ,2 To date, there are two TNFα blocking agents in clinical use, a monoclonal antibody (infliximab) and a soluble TNFα receptor (etanercept). The mechanisms behind the clinical effect of the TNFα blocking treatment are not fully understood, but both a reduced influx of inflammatory cells into the joints owing to downregulation of synovial endothelial adhesiveness3 ,4 and a direct interruption of the cytokine cascade driving the inflammatory process5 have been seen.
The association of RA with certain major histocompatibility complex class II genotypes has indicated that T cells are of importance in the pathogenesis. However, T cells obtained from either the circulation or from the synovial fluid of patients with RA are hyporesponsive when stimulated in vitro with antigen or mitogen compared with T cells from the blood of healthy controls.6-11 Earlier studies have shown that activated monocytes/macrophages suppress T cell functions, and this may be effected through the action of proinflammatory cytokines such as TNFα. Thus besides being an effector cytokine in the immune system, TNFα may also have immune modulating properties, something that has also been shown in animal studies.12Several studies have indicated that TNFα blockade with infliximab increases T cell reactivity.13 ,14 However, the influence of etanercept treatment has not been studied in this respect and there is also limited knowledge of how TNFα blockade influences immune reactivity against specific microbial agents or autoantigens. Against this background we wanted to investigate peripheral blood cytokine production and T cell reactivity to purified protein derivative (PPD), influenza virus, and the autoantigen collagen type II (CII) in patients with RA during etanercept treatment.
Methods
PATIENTS AND HEALTHY CONTROLS
Seventeen patients fulfilling the American College of Rheumatism (ACR) criteria for RA15 were recruited (12 women, five men, median age 48, range 31–69). Each patient had a history of unsuccessful treatment with at least one disease modifying antirheumatic drug (DMARD). The median disease duration was 132 months (range 24–324). Twelve patients (71%) were rheumatoid factor positive, 11 (65%) were treated with methotrexate. One patient received a combination of auranofin and methotrexate, another methotrexate and sulfasalazine, and one patient sulfasalazine alone. Twelve patients received prednisolone, either alone (n=4) or in combination with the DMARDs mentioned (n=8). In five patients auranofin, hydroxychloroquine, cyclosporin A in combination with mykophenolatmophetil (CellCept), sulfasalazine resp CPH-82 (Reumacon) were washed out two weeks before the start of etanercept treatment. No patient was treated with more than 10 mg prednisolone daily. Initially, the median C reactive protein was 34 mg/l (range 10–166).
Patients were treated with subcutaneous injections of 25 mg etanercept (Wyeth-Ayerst16) twice every week. Heparinised blood was collected before the start of treatment and after four and eight weeks of treatment. Healthy controls of similar age and sex as the patients with RA (median age 50, range 40–58, 12 female, five male) were recruited from the rheumatology laboratory and clinic at Karolinska Hospital.
EVALUATION OF CLINICAL RESPONSE
The clinical response of the treatment was measured in accordance with the ACR response criteria,17 using the 28 joint count.
CELL SEPARATION
Peripheral blood was collected in heparinised tubes and diluted 1:2 with phosphate buffered saline. Mononuclear cells were isolated by density gradient centrifugation and diluted to 1 × 106cells/ml in RPMI 1640 (Flow laboratories, Irvine, Scotland, UK) supplemented with glutamine, HEPES buffer, penicillin, streptomycin, and 10% of a defined batch of fetal calf serum (FCS) (Flow). These cells were then used without further stimulation for the enumeration of cytokine producing cells and for antigen and mitogen stimulation.
ELISPOT ANALYSIS OF NUMBER OF CYTOKINE PRODUCING PERIPHERAL BLOOD MONONUCLEAR CELLS
ELISA plates were coated with primary antibody, 50 μl/well, 15 μg/ml overnight at 4°C. Cells were added (105cells/well for interferon γ (IFNγ), 2.5 × 104/well for interleukin 10 (IL10), 2000/well for IL6, and 500/well for TNFα) in RPMI-10% FCS and incubated overnight. Wells were washed and a biotinylated antibody was added at 1 μg/ml overnight. Avidine-alkaline phosphatase (Dakopatts) was added at a dilution of 1:250 and allowed to bind for two hours, and 5-bromo-4-chloro-3-indolyl phosphate, 2.3 mM (Sigma) was added after washing to develop the ELISPOT for five hours. The number of cytokine producing cells was counted with an inverted-beam microscope. Antibodies used were 1-D1K (MabTech) and biotinylated 7-B6-1 (MabTech) for IFNγ, 19F1 (Pharmingen) and biotinylated 12G8 (American Type Culture Collection (ATCC), Manassas, VA, USA) for IL10, 13A5 (Pharmingen) and biotinylated 39C3 (Pharmingen) for IL6, and 20-A4 (ATCC) and biotinylated secondary antibody derived from Genzyme's Duokit for TNFα.
ANTIGEN AND MITOGEN STIMULATION OF PBMC
Antigen or mitogen (PPD 10 μg/ml (Statens Smittskyddsinstitut, Solna, Sweden), killed whole influenza virus diluted 1:1000 (Vaxigrip, Pasteur Mérieux, Lyon, France), chick CII 100 μg/ml (Sigma, St Louis, MO, USA), or phytohaemagglutinin (PHA) 5 μg/ml (Statens Smittskyddsinstitut)) were added to 1 ml of the cell suspension. Cells were incubated in round bottomed, 96 well plates (NUNC A/S, Roskilde, Denmark) and supernatants were collected and frozen after three days for the analyses of IL2 and PHA induced IFNγ and after seven days for the analyses of antigen induced IFNγ. The kinetics of cytokine production were analysed in pilot studies and found to correspond to a previous publication.18
CYTOKINE MEASUREMENTS IN SUPERNATANTS USING ELISA
Supernatants were analysed for cytokine content by ELISA using paired antibodies for IFNγ analysis (1-D1K as primary antibody and 7-B6-1 as biotinylated secondary antibody, MabTech, Stockholm, Sweden) as described earlier.6 For IL2 analysis, a commercial Duokit was used (R&D Systems, Minneapolis, MN, USA), as recommended by the manufacturer.
STATISTICAL ANALYSIS
Non-parametric methods were used throughout the report. Differences between groups were analysed with the Mann-Whitney U test, and analyses for matched pairs were performed with Wilcoxon's signed rank test. p<0.05 was considered significant.
Results
NUMBER OF CELLS SPONTANEOUSLY PRODUCING CYTOKINES IN PERIPHERAL BLOOD MONONUCLEAR CELLS
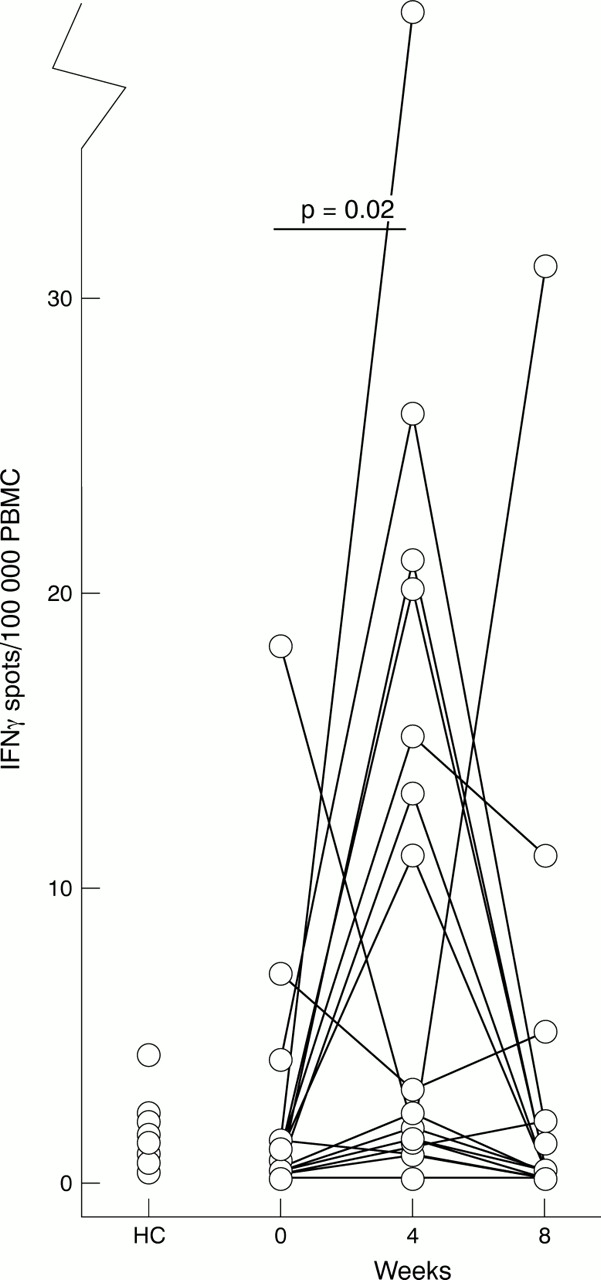
The number of cells producing IFNγ, IL10, IL6, and TNFα without further stimulation was determined by the ELISPOT method. All numbers given represent the number of cytokine producing cells/100 000 peripheral blood mononuclear cells (PBMC) as median and range. After four weeks of treatment with etanercept, a clear increase in the number of cells spontaneously producing IFNγ was recorded (fig 1). However, this effect was transient, as no difference between the number of cells producing IFNγ at baseline and after eight weeks of treatment was recorded. No difference in the number of IFNγ producing cells was seen between controls and patients with RA before etanercept treatment (fig 1).

The number of cells spontaneously producing interferon γ (IFNγ) increases after four weeks of treatment with etanercept. The number of IFNγ producing cells/100 000 unstimulated peripheral blood mononuclear cells (PBMC) was enumerated by the ELISPOT method. PBMC from healthy controls (HC, n=17) or patients with RA (n=17) were analysed at baseline and after four and eight weeks of treatment.
We detected a significantly increased number of TNFα producing cells in patients with RA at baseline (15 400 (range 6600–48 800)) compared with controls (12 000 (range 6800–22 000), p=0.04). However, etanercept treatment had no effect on the number of TNFα producing PBMC (data not shown).
There were no differences in IL10 and IL6 production between controls and patients with RA at baseline (data not shown). Etanercept treatment had no effect on the numbers of IL6 and IL10 producing cells (data not shown).
ANTIGEN REACTIVITY AFTER ETANERCEPT TREATMENT
A marked T cell hyporesponsiveness to microbe derived recall antigens was seen in patients with RA as compared with controls.6 ,10 This was evident for PPD induced IFNγ (fig 2A) and influenza induced IFNγ (fig 2C), as well as for influenza induced IL2 73 pg/ml (12–200) for controlsv 46 pg/ml (11–171) for RA, p=0.02, figure not shown).

In vitro antigen reactivity was increased after etanercept treatment. Peripheral blood mononuclear cells (PBMC) were separated from the blood of healthy controls (HC, n=17) or patients with RA (n=17) at baseline and after four and eight weeks of treatment. PBMC were incubated with the microbial derived antigen purified protein derivative (10 μg/ml) (A and B), influenza virus (C), or with collagen type II (100 μg/ml) (D). Supernatants were collected after seven days for the analysis of interferon γ (IFNγ; A, C, and D) and after three days for the analysis of interleukin 2 (IL2) (B). Cytokine measurements were performed using ELISA. Levels of cytokines obtained after incubation without further stimulation (or with acetic acid as a buffer control for collagen type II stimulation) were subtracted.
After four weeks of etanercept treatment, PPD-induced in vitro production of IFNγ (fig 2A) and IL2 (fig 2B), and influenza-induced in vitro production of INFγ (fig 2C) increased compared with baseline. After eight weeks of etanercept treatment, IFNγ responses, but not IL2 responses, showed a sustained increase compared with baseline.
Upon stimulation with CII, a cartilage derived antigen, no difference in the production of IFNγ could be detected between patients with RA and controls (fig 2D), confirming earlier published results from our laboratory6 and pointing towards a lack of T cell hyporesponsiveness to the autoantigen CII in patients with RA. The IFNγ response to CII stimulation increased after both four and eight weeks of treatment with etanercept compared with baseline (fig 2D).
MITOGEN REACTIVITY AFTER ETANERCEPT TREATMENT
PHA stimulation of control PBMC resulted in a significantly higher IFNγ response than that of RA PBMC at baseline (fig 3A), but we detected no difference between controls and RA in IL2 response to PHA stimulation (fig 3B). IFNγ production induced by PHA was not affected by etanercept treatment (fig 3A). However, the IL2 response to PHA stimulation was significantly increased after both four and eight weeks of treatment compared with baseline (fig 3B).

{kind=link}
{kind=link}
{kind=link}
In vitro mitogen reactivity was increased after treatment with etanercept. Peripheral blood mononuclear cells were separated from the blood of healthy controls (HC, n=17) or patients with RA (n=17) at baseline and after four and eight weeks of treatment and incubated with phytohaemagglutinin (5 μg/ml). Supernatants were collected after three days for the analysis of interferon γ (IFNγ) (A) and interleukin 2 (IL2) (B) using ELISA. Levels of cytokine obtained after incubation without further stimulation were subtracted.
CLINICAL RESPONSE
The clinical response to etanercept was in agreement with an earlier description from controlled clinical trials.19 ,20Thus 13 of the 17 patients (76%) fulfilled the ACR 20 criteria, seven patients (41%) also met the ACR 50 criteria, and one fulfilled ACR 70. Four of the 17 patients (24%) did not fulfil the ACR 20 response criteria.
No association was detected between response to treatment and any of the T cell functional variables measured. Neither could we distinguish any difference in T cell activity between the patients treated with methotrexate and the rest of the patient group. To investigate whether changes of drugs during the study period influenced the results, a subgroup analysis was made on patients that had stable drug treatment throughout the study. The results for IFNγ production and T cell reactivity were confirmed also in this subgroup (data not shown).
Discussion
This is the first report on functional studies of T cells from patients with RA treated with etanercept and we demonstrate an increase in peripheral T cell functions after treatment with the soluble TNFα receptor. This increase in T cell function was measured both as an increase in production of IFNγ of unstimulated PBMC and as an increase in production of IFNγ and IL2 after stimulation with microbial antigens (PPD or influenza) or autoantigen (CII) in vitro.
We chose to measure T cell activity using two different approaches. Firstly, we analysed IFNγ production ex vivo without further stimulation using the highly sensitive ELISPOT technique. We believe that this technique mirrors the cytokine production in vivo more accurately than other methods in which mitogenic stimulation is necessary for the enumeration of cytokine producing cells. Secondly, we determined the capacity for antigenic and mitogenic induction of IFNγ and IL2 by analysing cytokine secretion in cell culture supernatants using ELISA. We chose to study the microbial recall antigens PPD and influenza, as well as the cartilage derived antigen, CII. We also studied mitogen reactivity using PHA. Interestingly, we noted an increased general in vivo production of IFNγ and an increased in vitro responsiveness to both microbial antigens and to CII after treatment with etanercept. The in vitro data on reactivity to certain microbial antigens concord well with those published earlier on peripheral T cell function after treatment with infliximab.13 ,14 These types of data have not previously been documented after etanercept treatment, however, and data have also been lacking on whether TNFα blockade might enhance T cell reactivity to disease associated autoantigens like CII.
One mechanism by which TNFα blocking treatment might increase peripheral T cell function is by interfering with the TNFα dependent activation of monocytes. It is known that T cells from patients with RA can be functionally hyporesponsive9 ,11 ,21 as a result of the disease itself and irrespective of immunosuppressive drugs,10 possibly owing to the presence of activated monocytes during active inflammation. Activated monocytes suppress T cell functions by production of TNFα,13 ,22 ,23prostaglandins,24 and hydrogen peroxide.25Our determination of an increased production of TNFα in RA PBMC is in concordance with reports of a higher activation state of peripheral monocytes in RA than controls.26-28 These data support the notion of a monocyte induced and TNFα dependent downregulation of T cell functions in RA. By blocking the action of TNFα in vivo, it is conceivable that the activation of peripheral monocytes is decreased, as indicated by a decrease in serum levels of TNFα, IL1 receptor antagonist, IL6, and soluble TNFα receptor.29 This deactivation of peripheral monocytes might thereby result in an enhanced activation and reactivity of peripheral T cells.
Another possible mechanism behind the increased peripheral T cell reactivity noted after TNFα blocking treatment might be an increase in the number of circulating lymphocytes.5 ,14 This could be due to a decrease in ICAM-1 expression in synovial endothelial cells,3 ,4 which might reduce the extravasation of activated peripheral T cells. A decrease in locally produced TNFα might also affect cellular infiltration of the joint owing to the chemotactic property of TNFα.30 An increase of peripheral T cells with an activated phenotype,14resembling T cells normally present in the joint of patients with RA, further supports the idea of a change in T cell trafficking in patients treated with TNFα blocking agents. As inflammation is suppressed during treatment, the frequency of activated T cells declines, indicated by the reduced number of IFNγ producing cells in the peripheral blood at week 8. No difference in etanercept induced T cell activity was detected between patients responding to treatment and those without clinical response. This could be explained by individual differences in the cytokine regulation.31
Patients with RA have an increased mortality due to infections than healthy controls.32 The reason for this increased risk of infection may be the dysregulation of the immune system in RA, which suppresses the function of T cells. Immunosuppressive drugs used to control the disease have also been associated with a risk of infection.33 ,34 Although one might expect an even higher risk of infection when blocking TNFα, a cytokine of pivotal importance in protection against infectious agents, there is presently little published evidence supporting the increased presence of infections in patients with RA treated with etanercept.19 ,35 The findings of an increased T cell reactivity to recall microbial antigens after etanercept treatment may partly explain this paradox.
It has been postulated from several studies of experimental autoimmune diseases, such as lupus, experimental autoimmune encephalomyelitis, and diabetes, that blockade of TNFα might lead to exacerbation of disease under certain circumstances.12 ,36 ,37 To date, there is only one such report in multiple sclerosis that this theoretical concern may be of clinical significance,38 and results from clinical trials in RA using etanercept and infliximab have hitherto not corroborated that risk. However, there have been reports of the appearance of increased autoantibody production after infliximab treatment.2 ,39 ,40 It is thus interesting to note the increased T cell reactivity to CII in some of the patients treated with etanercept in this study. Although we do not know the exact clinical significance of this anti-CII T cell reactivity, our data suggest that increased reactivity of potential, disease inducing T cells may indeed also occur in patients treated with etanercept.
In conclusion, our data corroborate the notion that TNFα blockade may have stimulating effects on T cell reactivity against microbial agents, thereby possibly compensating for conceivable suppression of infection defence during TNFα blocking treatment. On the other hand, the stimulation of anti-self reactions in our study further emphasises the need for close supervision of patients receiving TNFα blockade so that potential unwanted effects of this new therapeutic principle may be recognised rapidly.
Acknowledgments
The work was supported by grants from the Swedish Medical Research Council and the Swedish Association against Rheumatism. We thank Associate Professor Robert A Harris for linguistic advice.
References
Footnotes
↵* Contributed equally.