Article Text

Abstract
Background Animal models of arthritis are frequently used to evaluate novel therapeutic agents. However, their ability to predict responses in humans is variable.
Objective To examine the time course of signalling molecule and gene expression in two models of arthritis to assist with selection of the model and timing of drug administration.
Methods The passive K/BxN serum transfer and collagen-induced arthritis (CIA) models were studied. Activation of MAP kinase and interferon (IFN)-response pathways was evaluated by quantitative PCR and western blot analysis of ankle joints at various time points during the models.
Results The kinetics of gene expression and kinase phosphorylation were strikingly different in passive K/BxN and CIA. All three MAP kinases (ERK, JNK and p38) and upstream kinases were activated within days in passive K/BxN and declined as arthritis severity decreased. Surprisingly, IFN-regulated genes, including IRF7, were not induced in the model. In CIA, activation of ERK and JNK was surprisingly low and p38 phosphorylation mainly peaked late in the disease. IFN-response genes were activated during CIA, with especially prominent peaks at the onset of clinical arthritis.
Conclusions Timing of treatment and selection of CIA or passive K/BxN might have an important impact on therapeutic response. p38, in particular, increases during the late stages of CIA. ERK and JNK patterns are similar in passive K/BxN and rheumatoid arthritis (RA), while IFN-response genes in CIA and RA are similar. The dichotomy between RA and animal models could help explain the poor correlation between efficacy in RA and preclinical studies.
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory disease marked by synovial inflammation and joint destruction.1 Although treatment of RA has improved in the past decade, there is still a need for new treatments. The use of preclinical models of arthritis has played a key part in drug development to help bridge this gap. Many animal models have been used to validate potential therapeutic targets. However, none are exact replicas of RA, even though they might share some pathogenic mechanisms.2 The accurate interpretation and use of rodent arthritis models, therefore, depends on understanding how they relate to RA.
Discrepant results in human and rodent studies are well known and were recently reviewed in detail.3 To improve the utility of the models in drug discovery, we profiled key molecules of the kinome and the interferon (IFN) response in murine arthritis systems that rely on innate immunity (passive K/BxN arthritis) or adaptive immunity (collagen-induced arthritis (CIA)). Our studies suggest that careful selection of models and the timing of treatment are critical variables that should be incorporated into target validation studies. By carefully designing these studies, the predictive value of preclinical experiments can potentially be increased and drug development expedited.
Methods
Mice
C57BL/6 and DBA1/J mice were purchased from The Jackson Laboratory (Bar Harbor, Maine, USA). All animal protocols received prior approval by the institutional review board.
Passive K/BxN arthritis
C57BL/6 recipient mice were injected with 100 µl of pooled adult K/BxN mice serum intraperitoneally on days 0 and 2.4 Clinical arthritis scores were evaluated using a scale of 0–4 for each paw for a total score of 16. Four injected mice and one control mouse were sacrificed at each time point. Arthritis scores and incidence for each time point include only the mice sacrificed for mRNA and protein analysis.
Collagen-induced arthritis
DBA1/J mice were immunised with bovine type II collagen (1 mg/ml) in complete Freund's adjuvant as previously described (Chondrex, Redmond, Washington, District of Columbia, USA).5 6 On day 21, 100 µg of bovine type II collagen in 0.1 ml of phosphate-buffered saline was injected intraperitoneally. On day 28, 5 µg of lipopolysaccharide 0111:B4 (Chondrex) in 0.1 ml of phosphate-buffered saline was injected intraperitoneally to synchronise disease onset and decrease variability that would complicate the kinetic analysis. Clinical arthritis scores were evaluated using a scale of 0–4 for each paw for a total score of 16. Six immunised mice and one control mouse were sacrificed at each time point. Arthritis scores and incidence for each time point include only the mice sacrificed for mRNA and protein analysis.
Antibodies
Anti-MKK6 antibody was purchased from Stressgen (Ann Arbor, Michigan, USA). Anti-P-Elk-1, anti-Elk-1 and anti-GAPDH antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, California, USA). All other antibodies were purchased from Cell Signaling Technology (Danvers, Massachusetts, USA).
Western blot analysis
Snap-frozen joints were pulverised and homogenised at 100 mg of tissue per 1 ml of lysis buffer. Western blot analysis was then performed on pooled samples from each time point as described previously.7 Immunoreactive protein was detected with Immun-Star Western C kit (Bio-Rad, Hercules, California, USA) using VersaDoc MP4000 imaging system (Bio-Rad). Densitometry analysis was carried out with Quantity One 1-D analysis software (Bio-Rad).
Quantitative real-time PCR
RNA derived from snap-frozen joint samples was isolated as described previously.8 Quantitative real-time PCR was performed with Assays-on-Demand gene expression products (Applied Biosystems, Foster City, California, USA).8 Sample threshold cycle (Ct) values were converted into cell equivalents of expression using a standard cDNA curve and normalised to the cell equivalents of Hprt1 expression to obtain relative cell expression units.
Statistical analysis
Data are expressed as mean ± SEM. Comparisons of quantitative PCR data were performed by Student t test with Bonferroni's correction, where appropriate. For western blot analysis, joint extracts for animals were pooled, which prevented formal statistical analysis.
Results
Kinetic profile of gene expression and signalling in passive K/BxN serum arthritis
Clinical arthritis
Administration of K/BxN serum caused severe arthritis in all the mice by day 4. Clinical scores increased through day 8 and declined by day 12 (see figure 1).
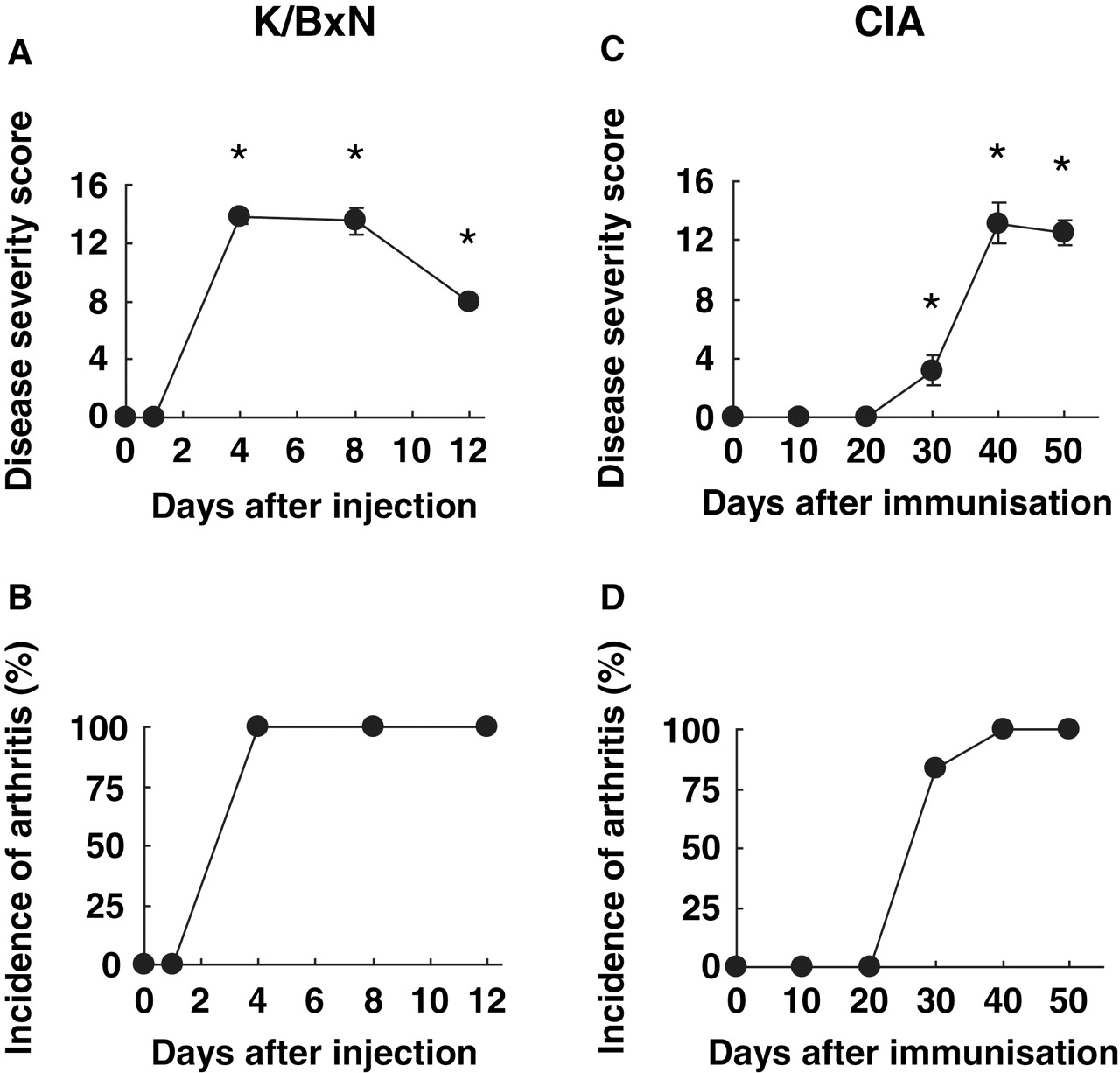
Disease severity scores in arthritis models. (A) Clinical arthritis scores for passive K/BxN arthritis. Values are the mean ± SEM (n=4/time point; one of three experiments shown). (B) Incidence of arthritis. (C) Clinical arthritis scores for collagen-induced arthritis (CIA). Values are the mean ± SEM (n=6/time point). (D) Incidence of arthritis. The percentage of mice with arthritis at each time point is shown.
Mediator gene expression
Initial experiments were performed to quantify gene expression in the joints of mice with passive K/BxN arthritis on days 0, 1, 4, 8 and 12 after serum administration. As shown in figure 2, interleukin (IL)6, IL10 and matrix metalloproteinase 3 expression increased within 1 day, which was before the onset of clinical arthritis. Expression of these genes remained high until day 8 and then decreased as disease severity declined. Tumour necrosis factor (TNF) expression was not increased, which is consistent with the observation that TNF has a relatively modest role in this model compared with IL1.9 Of the IFN-response genes, which are defined by the presence of an IFN-specific response element in their promoters and identified using microarrays,10 11 IFNβ and RANTES mRNA expression surprisingly decreased during the course of the model. IP-10, however, increased early in disease and later returned to baseline.

Synovial cytokine gene expression in passive K/BxN arthritis and collagen-induced arthritis (CIA). (A) Passive K/BxN arthritis. (B) CIA. Articular mRNA expression was determined by quantitative real-time PCR and normalised to Hprt1. Joint extracts were prepared for each mouse on the indicated days. Values are the mean ± SEM. REU, relative expression units (see ‘Methods’ section). *p<0.05 compared with control. n=4 for passive K/BxN arthritis and n=6 for CIA. IFN, interferon; IL, interleukin; MMP, matrix metalloproteinase; TNF, tumour necrosis factor.
MAP kinase pathway
To determine the kinetic profile of MAP kinases in the joints of mice with K/BxN serum-induced arthritis, western blot analysis was performed. Joint extracts from each selected day were pooled for evaluation of multiple signalling pathways simultaneously. Phosphorylation of all three MAP kinases (p38, JNK and ERK) increased within 1 day and peaked on day 4, which was when clinical scores reached their maximum level. JNK and ERK activation decreased on days 8 and 12 (figures 3, 4 and 5 for p38, JNK and ERK, respectively). However, P-p38 remained elevated even though clinical disease regressed.
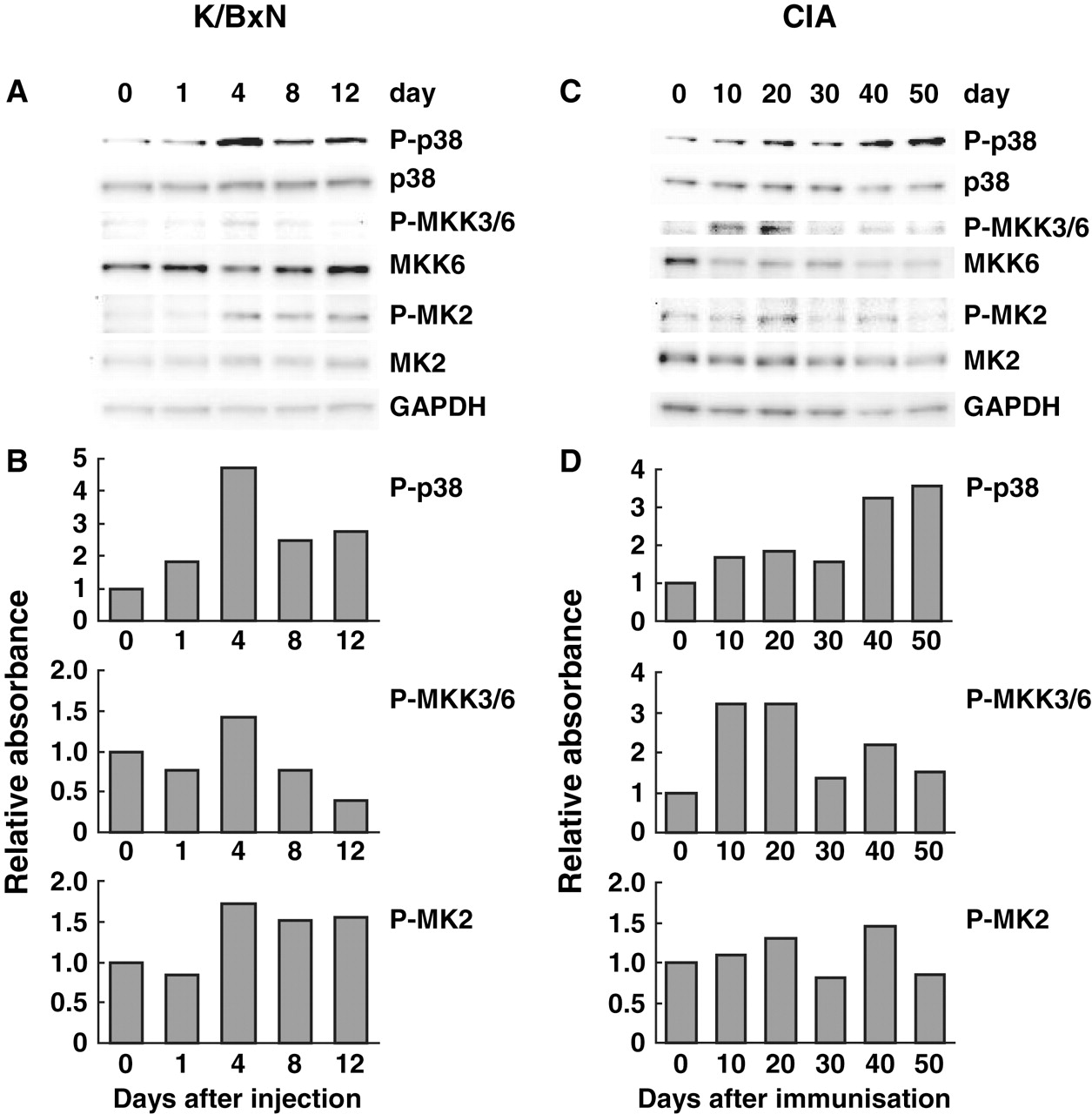
Kinetics of p38 pathway activation in joints of mice with passive K/BxN arthritis and collagen-induced arthritis (CIA). Joint extracts were prepared and pooled for each time point. This process allowed us to determine mean expression but precluded formal statistical analysis. (A, C) Phosphorylation of each MAP kinase, its upstream kinase and substrate were evaluated by western blot analysis. (B,D) Quantitative analysis of western blots (arbitrary densitometry units) after normalising results to GAPDH. n=4 for passive K/BxN arthritis and n=6 for CIA.

Kinetics of JNK pathway activation in joints of mice with passive K/BxN arthritis and collagen-induced arthritis (CIA). Joint extracts were prepared and pooled for each time point. (A,C) Phosphorylation of each MAP kinase, its upstream kinase and substrates were evaluated by western blot analysis. (B,D) Quantitative analysis of western blots (arbitrary densitometry units) after normalising results to GAPDH. n=4 for passive K/BxN arthritis and n=6 for CIA.

Kinetics of ERK pathway activation in joints of mice with passive K/BxN arthritis and collagen-induced arthritis (CIA). Joint extracts were prepared and pooled for each time point. (A,C) Phosphorylation of each MAP kinase, its upstream kinase and substrates were evaluated by western blot analysis. (B,D) Quantitative analysis of western blots (arbitrary densitometry units) after normalising results to GAPDH. n=4 for passive K/BxN arthritis and n=6 for CIA.
The MKKs are upstream kinases that regulate the MAP kinases. While there is some overlap in their specificity, ERK is usually activated by MEK1 and 2, p38 by MKK3 and 6 and JNK by MKK4 and 7.12,–,14 The kinetics of MKK phosphorylation was generally similar to that of the MAP kinases (figures 3–5). Late activation of P-p38 in the absence of P-MKK3/6 correlated best with P-MKK4 induction.15 P-MKK7 levels were not determined because the antibodies could not reliably detect this kinase in joint extracts (data not shown).
Activation of p38, JNK and ERK substrates was also examined. Phosphorylation of MK2, a p38α substrate, was similar to the kinetics of p38. Phosphorylation of c-Jun, which is one of the main substrates of JNK, peaked on day 8. P-Elk-1, which is a substrate of ERK, persisted until day 12 despite a reduction in P-ERK levels, possibly because other kinases can activate this transcription factor.
IRF pathway
Studies of the IRF pathway focused on mRNA because IKKε and IRF7 are transcriptionally regulated. Surprisingly, IRF3, IRF7 and IKKε gene expression either remained unchanged or decreased in the passive K/BxN model (see figure 6). These results correlated with the lack of RANTES and IFNβ induction (see figure 2).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Kinetics of IRF pathway activation in joints of mice with passive K/BxN arthritis and collagen-induced arthritis (CIA). Articular mRNA expression was determined by quantitative real-time PCR and normalised to Hprt1 for (A) passive K/BxN and (B) CIA. Joint extracts were prepared for each mouse on the indicated days. Values are the mean ± SEM. REU, relative expression units (see ‘Methods’ section). *p<0.05 compared with control. n=4 for passive K/BxN arthritis and n=6 for CIA.
Kinetic profile of gene expression and signalling in CIA
Clinical arthritis
Immunisation with type II collagen caused severe arthritis in all mice, with synovitis beginning around day 30, peaking on day 40 (see figure 1).
Mediator gene expression
The profile for gene expression in CIA is distinct from passive K/BxN arthritis (figure 2). IL6 expression increased markedly on day 30 but then decreased to near baseline. Matrix metalloproteinase 3 expression, like IL10, also increased by day 30 but persisted throughout, while TNF was mainly expressed early in the model. The profile of IFN-response genes also differed from passive K/BxN, with a narrow window of markedly increased RANTES expression on day 30. IP-10 mRNA levels increased early but declined to baseline. As with the K/BxN experiment, IFNβ expression was below baseline at most time points.
MAP kinase pathway
p38 activation increased modestly during the early phases of the model, with a peak on day 20 (figure 3). However, the major increase was delayed until when arthritis was well established. The JNK pathway was striking in that only the 54 kDa isoform was activated early in the model (figure 4). Surprisingly, P-ERK did not increase during CIA (figure 5).
MKK activation was also seen in CIA (figures 3–5). Of interest, P-MKK3/6 peaked early, which was temporally related only to the early P-p38 peak. P-MKK4 increased later in the model and correlated with delayed P-p38 expression. As with passive K/BxN, MKK4 and JNK activation did not correlate. MEK1/2 activation, like ERK, was not increased in CIA.
MAP kinase substrate activation in CIA was surprisingly modest (figures 3–5). c-Jun phosphorylation increased marginally. However, when P-c-Jun was normalised to total c-Jun instead of GAPDH, relative P-c-Jun levels increased by about fourfold (data not shown). P-MK2 increases were quite modest, but occurred at the same time points as the P-p38 peaks.
IRF pathway
Activation of the IRF pathway in CIA correlated with IFN-response gene expression (see figures 2 and 6). IRF7 increased in the same window frame as RANTES. IKKε gene expression, which did not change in the passive model, was higher throughout CIA and peaked on day 30 when RANTES expression also was maximal. IRF3 expression was unchanged throughout the experiment, with a trend towards lower levels during CIA.
Discussion
Mouse models of inflammatory arthritis are widely used to assess the utility of novel therapeutic agents. These models share many features with RA, but also differ from the human disease with respect to pathogenesis, relative contribution of innate and adaptive immunity and diverse responses to therapeutic interventions.2 3 16 The interpretation and rational use of preclinical models requires an understanding of each individual system and how it relates to RA, which is a heterogeneous disease that can involve multiple pathogenic mechanisms. This study was designed to profile the kinetics of several key signalling pathways in murine arthritis, an approach that we previously used to evaluate the time course of AP-1 and NF-κB expression in CIA.5 This information can be used to help determine the best ways to evaluate drugs in preclinical studies, including the model selection and the timing of drug administration (see table 1 for a summary of results).
Relative expression of genes and signalling molecules in passive K/BxN arthritis and collagen-induced arthritis
Mouse passive K/BxN and active CIA were chosen because they are widely used and have distinct mechanisms. Passive K/BxN arthritis is an antibody-induced arthritis that is dependent solely on innate immunity, including complement, neutrophils, mast cells and macrophages.17,–,19 In contrast, adaptive immunity participates in the pathogenesis of CIA through the generation of an antigen-specific response to type II collagen.20
MAP kinase inhibitors have been extensively studied in preclinical models of arthritis.21 Despite abundant evidence that p38 inhibitors are effective in mice and rats, these observations have not yet translated to efficacy in humans.22 Our profiling studies demonstrate that articular P-p38 levels generally correlate with clinical arthritis and proinflammatory mediator expression in early passive K/BxN arthritis and CIA. However, a second wave of P-p38 induction occurs during the late stages of both models when disease stabilises or regresses. Early p38 activation was also associated with phosphorylation of its two main upstream kinases—namely, MKK3 and MKK6. However, P-MKK3/6 induction was not observed in late disease. Instead, the MAP kinase kinase associated with P-p38 at these time points is MKK4, which can also activate p38.15 These observations suggest that p38 might possess anti-inflammatory functions in established disease that are regulated by MKK4 rather than MKK3/6. Of interest, anti-inflammatory activity has been associated with p38, especially through the production of IL10.23 The fact that P-p38 levels do not correlate with IL6 in CIA also suggests that the kinase might not tightly control some proinflammatory cytokines, especially in the late phases.
JNK was previously implicated as a therapeutic target in RA.24 We observed that the patterns of JNK phosphorylation are distinct in passive K/BxN arthritis and CIA. Both major molecular weight species (54 kDa and 46 kDa) are phosphorylated in the former and correlate with synovitis and proinflammatory gene expression. However, only the high molecular weight protein was activated in CIA, with a peak on day 30 when many proinflammatory genes are activated. MKK4, which can phosphorylate JNK, was not induced during early phase of CIA and MKK7 might be more important.25
The functions of individual JNK proteins vary depending on the cell lineage and mode of stimulation. The JNK family consists of JNK1, JNK2 and JNK3, each of which have multiple 54 kDa and 46 kDa isoforms due to alternative splicing.26 Despite their biochemical and structural similarities, JNK1 and JNK2 exert distinct effects on c-Jun turnover and activity.27 JNK2 is the major isoform of the JNK family expressed by human fibroblast-like synoviocytes.28 The 54 kDa and 46 kDa proteins are equally phosphorylated in cytokine-stimulated synoviocytes and RA synovium. This pattern suggests that passive K/BxN arthritis, rather than the CIA, might reflect JNK activation in RA and could be a better model for assessing JNK inhibitors.
One striking finding in our study was the lack of P-ERK and P-MEK1/2 in CIA compared with passive K/BxN arthritis. In contrast, P-Elk-1 was observed in both cases, probably because other kinases like JNK or p38 can phosphorylate this transcription factor.29 Some ERK and MEK1/2 inhibitors are beneficial in CIA, which might be due to effects on central immunity rather than the synovium.30 31 Because P-ERK is expressed in RA synovium,32 the ERK pathway in RA appears to correlate better with passive K/BxN arthritis than CIA.
The IFN signature has been implicated in many autoimmune and inflammatory diseases, including RA and systemic lupus erythematosus.33 34 The role of this pathway is complex; preclinical studies support the use of IFNβ itself as a therapeutic agent while other suggest that blocking IFN-response genes with the IFN-specific response element in their promoters, like RANTES and IP-10, might be beneficial.35 Therefore, we also evaluated the kinetics of IKKε, IRF3 and IRF7 gene expression in the joints of mice with passive K/BxN arthritis and CIA. IKKε is an IKK-related kinase that regulates IFN-mediated signalling and IFN-induced gene expression through IRF3 and IRF7 phosphorylation.36 We measured mRNA expression for these genes rather than protein because the IKKε and IRF7 genes are induced by inflammation in addition to post-translational modification.37,–,41
We were surprised to see either no change or a trend towards decreased IKKε and IRF7 expression in passive K/BxN arthritis. In contrast, the IRF pathway and IFN-regulated genes were highly activated in CIA. In some cases, as with IRF7, there was a spike in gene expression on day 30, a time when arthritis is accelerating. IRF7 expression correlated with synovial RANTES, both of which are regulated by IKKε.42 As IKKε-deficient mice have a modest reduction in the severity of arthritis in passive K/BxN arthritis, IKKε might play a part in synovial innate immune responses.43 Alternatively, the mechanism might be due to an effect on central immunity with an indirect effect on joints. IKKε is highly expressed and activated in RA synovial tissue, suggesting that CIA might be a more suitable model for evaluating IFN responses.44
In conclusion, this study demonstrates distinct kinetic profiles of the MAP kinase families and IFN-response molecules in models of arthritis. By tailoring studies to the mechanism of drug action, the signalling pathways and the relationship between murine and rheumatoid synovial inflammation, a rational approach to preclinical models in drug testing can be developed. This process can potentially limit the number of models that need to be evaluated and enhance our ability to interpret the results.
Acknowledgments
We thank Meghan Edgar and Katharyn Topolewski for expert assistance.
References
Footnotes
-
Funding These studies were supported, in part, by NIH grants R01AI070555, R01AI067752 and R01AR47825.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.