Article Text

Abstract
OBJECTIVE To investigate the thrombin inhibitory capacity of antithrombin III in the inflamed human joint.
METHODS Thrombin inhibitory capacity was measured, using a kinetic spectophotometric method, in matched plasma and synovial fluid samples of patients with rheumatoid arthritis (n=22) and osteoarthritis (n=16), together with normal control plasma samples (n=13). In the same samples, the concentration of antithrombin III was also determined by the method of radial immunodiffusion. The combination of these measurements allowed the calculation of the specific thrombin inhibitory capacity of these samples.
RESULTS An increased concentration of antithrombin III in rheumatoid compared with osteoarthritic synovial fluid was noted (p<0.05). However, there was a significant depression in the specific activity of antithrombin III in rheumatoid synovial fluid when compared with matched plasma samples (p<0.001) or with osteoarthritic synovial fluid (p<0.05).
CONCLUSION In rheumatoid synovial fluid the thrombin inhibitory capacity of antithrombin III is disproportionately depressed relative to the concentration of antithrombin III, indicating the inactivation of antithrombin III in the rheumatoid joint.
- antithrombin III
- thrombin
- rheumatoid arthritis
- synovial fluid
Statistics from Altmetric.com
Thrombin is a multifunctional enzyme that plays a central part in haemostasis and coagulation. It cleaves fibrinogen to form the fibrin clot and can activate factors V, VIII, XIII, and protein C and is a potent stimulator of platelet aggregation. Aside from these functions, thrombin has pro-inflammatory properties12 with a number of effects on the vascular endothelium, including stimulation of neutrophil adhesion to the vessel wall and of prostacyclin release through upregulation of arachidonic acid. Thrombin also acts as a mitogen for synovial cells1 and there is increased thrombin receptor expression by cells within the rheumatoid synovium.3 Fibrin deposition is a prominent finding within the rheumatoid synovium1 and hyperfibrinogenaemia is associated with rheumatoid arthritis (RA).
The most important physiological inhibitor of thrombin is antithrombin III (AT III).1 This single chain glycoprotein of molecular weight 58 000 is synthesised primarily in the liver and belongs to the serpin (serine proteinase inhibitor) superfamily. Serpins display sequence homology, share common structural features, and form a 1:1 complex with their cognate proteinase. A common structural feature of serpins is an exposed polypeptide loop that contains the reactive centre involved in proteinase inhibition. In the case of AT III, this is centred on Arg 393. The rate of thrombin inhibition by AT III is greatly enhanced by the presence of glycosaminoglycans, such as heparin.
In vitro, serpins are susceptible to the inactivation of their proteinase inhibitory activity by proteolytic attack at an exposed loop of the polypeptide chain (which contains the reactive centre) and/or oxidative inactivation involving reactive oxygen/nitrogen species.4 Both metalloproteinases and reactive oxygen species are thought to contribute to biomolecular damage within the RA joint.4 Previous studies have shown a decreased concentration of AT III in RA synovial fluid compared with RA plasma5 but the concentration of AT III protein has not been related to the thrombin inhibitory capacity of these fluids to obtain the specific activity of synovial fluid AT III. In vitro, ATIII is proteolytically inactivated by metalloproteinases4 and by neutrophil elastase.6 We therefore hypothesised that in RA, AT III may be inactivated, thus potentiating the pro-inflammatory actions of thrombin.
Method
Matched samples of plasma and synovial fluid were obtained from 38 patients. The 22 patients (16 female, 6 male) with RA and mean age of 64 years (range 45–84) satisfied the 1987 ARA revised criteria.7 The 16 patients (9 female, 7 male) with osteoarthritis (OA) of the knee and mean age of 71 years (range 54–81) were diagnosed on combined clinical and radiological grounds, which included pain on joint use and the presence of joint space narrowing with osteophytosis in at least one tibiofemoral compartment of the knee joint in all cases. Plasma was also obtained from 13 healthy subjects (7 female, 6 male) with a mean age of 36 years (range 21–53). All samples were collected in EDTA, centrifuged at 1500 g for 10 minutes, and immediately frozen at −70°C until analysis.
All of the patients with RA were receiving disease modifying drugs or non-steroidal anti-inflammatory drugs, or both, while most of the OA patients were receiving either no drug or paracetamol. No patients were receiving corticosteroids, nor had any received corticosteroids within 12 weeks of the study. RA activity was assessed by erythrocyte sedimentation rate (ESR), C reactive protein (CRP), and Ritchie index measurements.
Samples were assayed for thrombin inhibitor capacity (TIC) by a kinetic spectrophotometric method,8 using the Coatest Antithrombin kit (Quadratech, Epsom, Surrey). Test plasma or synovial fluid was diluted in buffer containing EDTA. An excess of heparin and thrombin was added. This was incubated for 30 seconds at 37°C and residual thrombin determined by adding a chromogenic substrate (S-2238) and polybrene to neutralise the heparin during the period of substrate hydrolysis. The cross reactivity of S2238 with plasma kallikrein was insignificant at the concentration of thrombin and kallikrein present in our assay system. This hydrolysis was monitored on a spectrophotometer at 405 nm as the initial reaction rate. The kit measured the activity of AT III as a percentage of normal plasma. Values were expressed relative to a reference plasma standard that had an activity of 0.95 IU/ml (NIBSC, Potters Bar, Herts). All percentage values were converted to IU/ml.
The concentration of AT III protein in plasma and synovial fluid was quantified immunochemically by radial immunodiffusion using NOR-Partigen Antithrombin III plates (Behring, Hounslow, Middx). The plates contain monospecific antiserum to human antithrombin III in a ready to use agarose gel layer. Plasma and synovial fluid samples were pipetted into the wells of the plate and allowed to incubate for 48 hours at room temperature. After the diffusion period the diameters of the precipitates were measured. The concentration of AT III was measured as a percentage of normal plasma, with the calibration table provided giving corresponding results in mg/ml.
By dividing the TIC of AT III in a sample by the immunochemically determined concentration of AT III protein, the specific thrombin inhibitory capacity (STIC) of AT III in that sample was calculated (IU/mg).
Statistical analysis was by Mann-Whitney U test and Spearman’s rank correlation. Results were expressed as the median and range. A p value of less than 0.05 was considered to be statistically significant.
Results
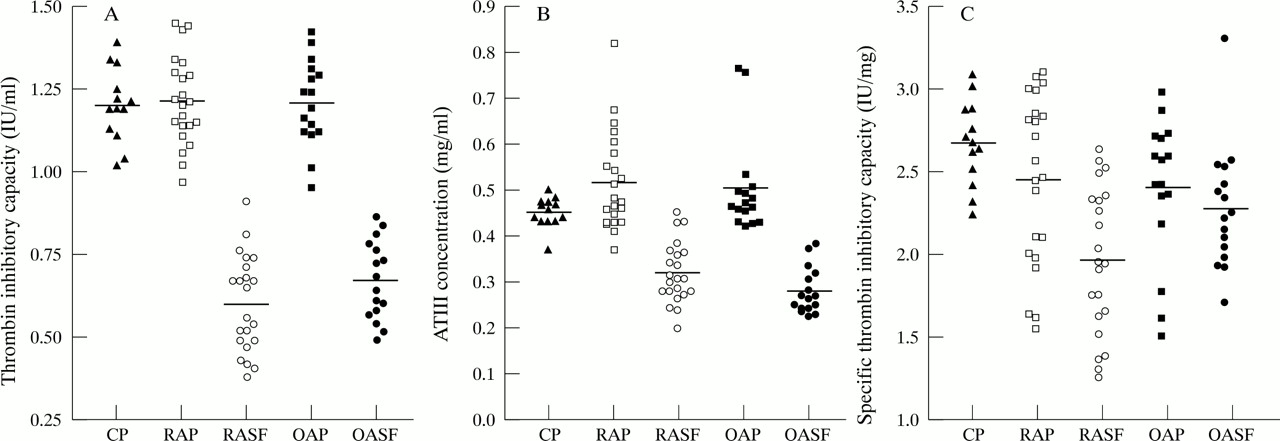
The median TIC (fig 1(A)) of RA and OA synovial fluid samples was significantly decreased when compared with respective plasma samples (p < 0.001). There was no significant difference between the TIC of RA and OA synovial fluid. The median concentration of AT III (fig 1(B)) was significantly lower in both RA and OA synovial fluid samples when compared with paired plasma samples (p < 0.001), while the AT III concentration was significantly higher in RA than OA synovial fluid (p < 0.05).
The STIC of AT III was comparable in the plasma of healthy subjects, RA, and OA patients. There was a significant decrease in the STIC of AT III (fig 1(C)) in synovial fluid from RA patients (p < 0.001), but not from OA patients, when compared with matched plasma. Furthermore, there was a significant depression in the specific activity of AT III in RA synovial fluid compared with OA synovial fluid (p < 0.05). The median percentage inactivation of AT III in RA synovial fluid, relative to normal plasma (defined as 100% active) was 32%, with a range of 1 to 53% inactivation.
There was no correlation between rheumatoid plasma and synovial fluid TIC and AT III concentrations and neither TIC nor AT III concentration correlated with ESR, CRP, Ritchie index or disease duration. There was no obvious relation between the drugs that patients were receiving and either TIC, AT III concentration or STIC. The healthy control group, RA and OA groups were analysed further, by age, sex, and smoking habit with no statistical difference noted in TIC, AT III concentrations or STIC.
Discussion
This study shows that the TIC of RA synovial fluid is disproportionately depressed relative to the immunochemically determined concentration of AT III protein. There is also a selective depression in the STIC of rheumatoid synovial fluid compared with OA synovial fluid. This suggests that a proportion, but not all, of the ATIII in RA synovial fluid has been inactivated. Possible mechanisms by which the inactivation of AT III may occur are discussed below, although other potential mechanisms undoubtedly exist.
COMPLEX FORMATION BETWEEN AT III AND THROMBIN
Concentrations of the thrombin-AT III complex are significantly increased in the synovial fluid from patients with juvenile chronic arthritis9 and in RA2 ,10 when compared with OA and cadaveric synovial fluid. Factor Xa-AT III complexes are also present at higher concentrations in rheumatoid as compared with OA and cadaveric synovial fluid.2 These increased levels of enzyme inhibitor complexes imply activation of coagulation within the RA joint. However, the concentration of thrombin-AT III complexes in rheumatoid synovial fluid is small, being less than 3% of the concentration of AT III, and the concentration of Factor Xa-AT III complexes is probably even lower.2 This suggests that other mechanisms are involved in AT III inactivation.
PROTEOLYSIS BY METALLOPROTEINASES/NEUTROPHIL ELASTASE
There is evidence for an increased activity within the RA joint of neutrophil elastase as well as metalloproteinases, including collagenase, gelatinase, and stromelysin, which are released from a variety of cells, including synovial fibroblasts.4 These enzymes not only degrade joint tissues, but also inactivate serpins by proteolysis within the exposed loop region referred to earlier. As mentioned earlier AT III can be inactivated by both metalloproteinases4 and neutrophil elastase.6To our knowledge, no direct evidence exists for the presence of proteolytically fragmented forms of AT III in RA synovial fluid, although this would seem to be one likely mechanism of AT III inactivation at this site.
OXIDATION BY REACTIVE OXYGEN/NITROGEN SPECIES
Serpins, even those that do not have a methionine residue at their reactive centre, such as PAI-1 are susceptible to oxidative inactivation.4 Again, we are unaware of any reports of the oxidation status of AT III in RA synovial fluid. However, lipid peroxides, which are present in RA synovial fluid4 have been shown to lower the thrombin neutralising activity of AT III.11 In this study, the inactive portion of AT III in RA synovial fluids was similar to that for α1AT in RA synovial fluid.4 As in the case of α1AT, however, the proportion of inactive AT III may be much higher within certain “micro-environments” of the joint. For example, within the extracellular space between the surface of oxidant and proteinase producing adherent macrophages and the cartilage surface. Weinberg et al noted a correlation between macrophage content and synovial fibrin deposition in patients with RA.1 Such microenvironments may explain the presence of fibrin deposits within the rheumatoid synovium.
PRESENCE OF AN INHIBITOR OF COMPLEX FORMATION BETWEEN THROMBIN AND ANTITHROMBIN III
Hogg and Jackson12 have shown that fibrin monomers reduce the susceptibility of thrombin to inactivation by AT III. Either fibrin monomers or other inhibitors in RA synovial fluid might block the binding of thrombin to antithrombin III.
In conclusion, whatever the mechanisms involved, our study shows a significant proportion of inactive AT III in RA synovial fluid. This may explain the intra-articular coagulation through decreased inhibition of thrombin, resulting in increased fibrin production in the rheumatoid joint. Both thrombin1 and fibrin13have pro-inflammatory effects and thrombin receptors are upregulated within the rheumatoid synovium.3 In a previous study the concentration of thrombin, but not AT III, in rheumatoid synovial fluid correlated directly with both ESR and CRP.10 AT III levels are augmented by anabolic steroids, progesterone, and oral anticoagulants.14 This may, in part, explain the effectiveness of corticosteroids in RA and raises the possibility of controlling intra-articular activation of coagulation as a treatment strategy for RA. Belch et al noted an increase in fibrinolysis and improved disease control in RA patients treated with stanozolol.15 Other possible treatments could include AT III concentrates, recombinant human AT III, hirudin, synthetic low molecular weight heparin or fibrinolytic agents. In practice, such approaches may be challenging because anticoagulation might predispose the patient to bleeding, including intra-articular haemorrhage, a well known pro-inflammatory stimulus.4
References

{kind=link}
Scatter plots showing the thrombin inhibitory capacity of antithrombin III and the antithrombin III concentration in paired RA and OA plasma and synovial fluid. (A) Thrombin inhibitory capacity. RA synovial fluid compared with RA plasma, p < 0.001; OA synovial fluid compared with OA plasma, p < 0.001. (B) Concentration of antithrombin III. RA synovial fluid compared with RA plasma, p < 0.001; RA synovial fluid compared with OA synovial fluid, p < 0.05. (C) Specific thrombin inhibitory capacity. RA synovial fluid compared with OA synovial fluid p < 0.05. CP; healthy control plasma, RAP; rheumatoid arthritis plasma, RASF; rheumatoid arthritis synovial fluid, OAP; osteoarthritis plasma, OASF; osteoarthritis synovial fluid. The central solid line represents the median.