Article Text

Abstract
Objectives To evaluate the efficacy and safety of adalimumab in patients with peripheral spondyloarthritis (SpA) not fulfilling the criteria for ankylosing spondylitis (AS) or psoriatic arthritis (PsA).
Methods 40 patients with active peripheral SpA fulfilling the European Spondyloarthropathy Study Group or Amor criteria but not the criteria for AS or PsA were included in a randomised, double-blind, placebo-controlled clinical trial. Patients were treated 1 : 1 with adalimumab or placebo for 12 weeks, followed by an open label extension up to week 24. Safety and efficacy measurements were performed every 6 weeks, with the patient's global assessment of disease activity at week 12 as the primary endpoint.
Results At week 12, the patient's and physician's global assessment of disease activity, swollen joint count, Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), Ankylosing Spondylitis Disease Activity Score (ASDAS) and erythrocyte sedimentation rate improved significantly in the adalimumab group compared with the baseline values and compared with placebo. A similar improvement was seen upon adalimumab treatment from weeks 12 to 24 in the patients originally randomised to placebo, whereas the clinical response was maintained or even augmented at week 24 in the patients who received adalimumab from the start. ASDAS inactive disease and BASDAI50 responses were met in 42% of the adalimumab group versus 0%–5% in the placebo group at week 12 (p=0.001 and p=0.008, respectively), and were further increased at week 24. The number of adverse events was not different between the adalimumab and placebo groups.
Conclusions Adalimumab appears to be effective and well tolerated in SpA patients with peripheral arthritis, also in those patients not fulfilling the AS or PsA criteria.
- Anti-TNF
- Spondyloarthritis
- Arthritis
- Treatment
Statistics from Altmetric.com
Introduction
The treatment of spondyloarthritis (SpA) has improved since the successful introduction of tumour necrosis factor (TNF)-blockade in this disease more than 10 years ago.1–5 However, this therapy is only approved and reimbursed for ankylosing spondylitis (AS) and psoriatic arthritis (PsA), which are the best described and studied phenotypic subtypes of SpA. Approximately a third of the SpA population cannot be classified as AS or PsA and belong to one of the other SpA subtypes: inflammatory bowel disease related SpA, reactive arthritis and undifferentiated spondyloarthritis.6 ,7 The last consists of patients with peripheral disease as well as patients with axial disease not fulfilling the modified New York criteria for AS,8 now commonly called non-radiographic axial SpA (nr-axSpA). Based on the similarities in symptoms and disease severity between nr-axSpA and AS,9 new classification criteria have been developed for axial SpA without phenotypic subclassifications.10 Supporting the concept that nr-axSpA is not fundamentally different from AS, several small placebo-controlled randomised clinical trials (RCTs) have demonstrated the efficacy of TNF-blockers in nr-axSpA.11–13 This has recently been confirmed in a large RCT with adalimumab.14
As for axial SpA, familial aggregation studies,15–17 synovial immunopathology18–20 and experimental models21 strongly suggest that also peripheral disease in SpA belongs to a single pathological entity. Accordingly, unifying criteria have recently been developed for peripheral SpA.22 TNF-blockade has a proven beneficial effect on peripheral manifestations of AS and PsA.3 ,4 ,23–26 In non-AS and non-PsA peripheral SpA, several small studies suggest a good efficacy of anti-TNF-therapy but RCTs are still lacking.27–32 Therefore, the objective of this study was to assess the efficacy and safety of adalimumab in a randomised, double-blind, placebo-controlled clinical trial in patients with peripheral SpA not fulfilling the criteria for AS or PsA.
Methods
Study design
Overall, 40 patients were randomised (1 : 1) in a single centre, double-blind clinical trial to receive adalimumab 40 mg or placebo subcutaneously every other week for 12 weeks, followed by an open label extension with adalimumab for another 12 weeks. Written informed consent was obtained from each patient before study related procedures were performed. If eligible, patients entered the study within 3 weeks. The primary endpoint of the study was the improvement in the patient's global assessment of disease activity at week 12. This endpoint was chosen because most other disease activity measurements in SpA are validated in AS only and because it has been used previously as primary endpoint in anti-TNF trials in mixed SpA populations.4 The study drug was provided in prefilled syringes containing adalimumab 40 mg or matching placebo (Abbott Laboratories, Abbott Park, Illinois, USA). The study was approved by the local Ethics Committee and is registered under the code NTR1806.
Patients
Patients had to fulfil the European Spondyloarthropathy Study Group (ESSG) criteria6 and/or the Amor criteria33 for SpA for at least 3 months without fulfilling the criteria for AS8 or PsA.34 The Assessment of SpondyloArthritis International Society (ASAS) criteria for peripheral SpA22 were not used as they were not published yet when the study was started. Patients had to be between 18 and 70 years old and had to have an active arthritis (at least one swollen and tender joint) despite treatment with non-steroidal anti-inflammatory drugs (NSAIDs). For female subjects, a negative pregnancy test and adequate contraception during the study and for 150 days thereafter were criteria for entry. Exclusion criteria included serious infections in the previous 4 weeks, history of malignancy in the past 10 years, significant history of other severe diseases or uncontrolled concomitant disease. Chest radiography and a tuberculin skin test were performed at screening. In case of active tuberculosis, patients were excluded and in case of latent tuberculosis, patients had to receive at least 3 months of isoniazide before enrolment.
Concurrent and prior medication
Patients were allowed to continue NSAIDs, corticosteroids (≤10 mg/day prednisone or equivalent), methotrexate and sulphasalazine provided that the dosage was stable for at least 4 weeks prior to baseline and throughout the study and, for methotrexate and sulphasalazine, provided they had been started at least 3 months before inclusion. Intra-articular corticosteroids and other disease-modifying antirheumatic drugs (DMARDs) were not allowed and were discontinued at least 4 weeks prior to baseline. Prior anti-TNF-therapy or investigational drugs were permitted if stopped more than 2 months prior to baseline.
Clinical response
Patients were seen for clinical evaluation at baseline, weeks 6, 12, 18 and 24 where the following disease activity parameters were evaluated: patient's and physician's global assessment of disease activity (each a 100 mm visual analogue scale), 68/66 tender joint count (TJC) and swollen joint count (SJC), Bath Ankylosing Spondylitis Disease Activity Index (BASDAI),35 Ankylosing Spondylitis Disease Activity Score (ASDAS),36 modified Schober index, erythrocyte sedimentation rate (ESR) and C reactive protein (CRP). Improvement at weeks 12 and 24 was also measured by the different ASDAS improvement criteria and BASDAI50 response.36–38 The Health Assessment Questionnaire Disability Index (HAQ-DI)39 and Health Utility Index Mark 3 (HUI-3)40 ,41 were measured as disability and health related quality of life (QoL) variables. At each visit patients were asked for side effects, and routine laboratory testing and physical examination were performed for safety evaluation.
Statistical analysis
The sample size calculation was based on previous anti-TNF trials which included peripheral SpA1 ,4 ,27 with an estimated response in patient's global assessment at week 12 of −48±24 mm in the adalimumab group and −21±32 mm in the placebo group, a power of 80% and α level of 0.05. The data were tested for normal distribution and presented as mean±SD or SEM. Analysis of covariance (ANCOVA) adjusting for the baseline score was used to compare change from baseline at week 12 between the adalimumab and placebo groups. Further comparison of the adalimumab and placebo groups was done by independent sample t test for continuous variables and χ2 test for categorical variables. Changes from baseline (intragroup analysis) were assessed by paired t tests. All statistical tests were two-sided and p values of <0.05 were considered statistically significant.
Results
Clinical and demographic characteristics at baseline
In all, 40 patients were enrolled in the study and subsequently randomised to treatment with adalimumab or placebo. A total of 31 patients fulfilled both the ESSG and Amor criteria, six only the ESSG criteria, and three only the Amor criteria. The demographic and clinical characteristics were similar across both treatment arms (table 1). Only human leukocyte antigen B27 (HLA-B27) positivity tended to be higher in the adalimumab group (55%) versus the placebo group (25%) (p=0.053).
Baseline characteristics of the study population by treatment group
At baseline, both treatment groups were well matched for all disease activity parameters except for the physician's global assessment, which was somewhat higher in the placebo group (57.0±12.6 mm) compared with the adalimumab group (47.8±11.8 mm) (p=0.022) (table 1). The mean SJC66 tended to be slightly higher in the patients who were assigned to adalimumab (4.3±4.2) compared with placebo (2.5±1.9) (p=0.096). The number of patients with elevated CRP (n=7 and n=8 in the adalimumab and placebo groups, respectively) and ESR (n=3 and n=4, respectively) at baseline was similar in the two groups.
Concomitant NSAID and DMARD use was not different between the treatment groups. One patient randomised to receive adalimumab and two patients randomised to receive placebo had previously been treated with TNF-blockers (infliximab or adalimumab) for inflammatory bowel disease, but in all cases TNF-blockade was stopped more than 1 year prior to baseline. Two patients tested positive for latent tuberculosis and were treated with isoniazide before entering the trial.
Improvement of disease activity at week 12
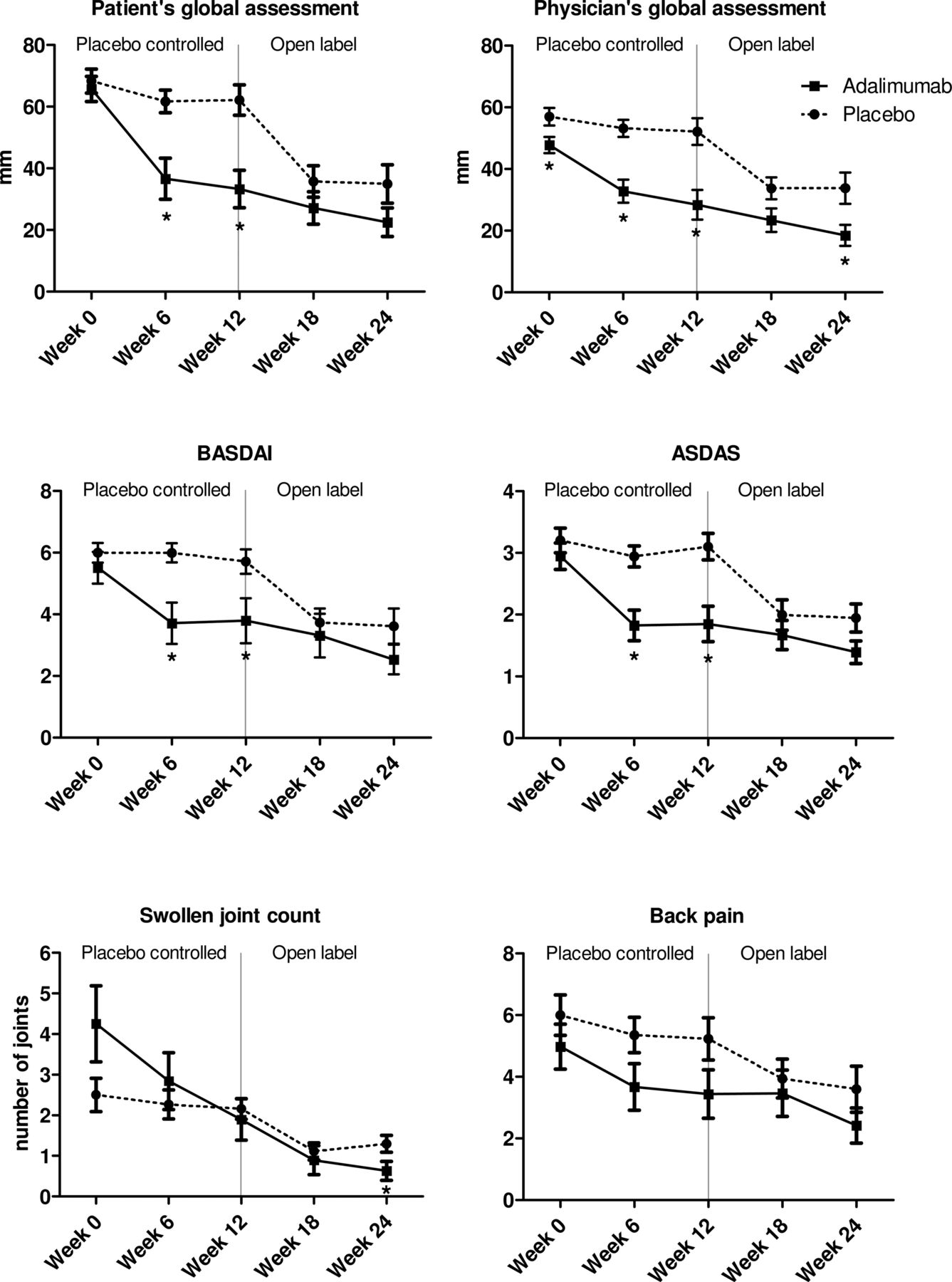
The primary endpoint of the study was met since the patient's global assessment showed a strong improvement at week 12 compared with baseline in the adalimumab group (−31.0±23.3 mm), while the placebo group showed almost no improvement (−5.9±21.4 mm) (p=0.001). Also the improvement in the physician's global assessment, BASDAI, ASDAS and ESR was significantly greater in the adalimumab treated patients compared with placebo (p<0.05 for all parameters) (table 2). These data were confirmed by direct comparison of the adalimumab and placebo groups: the patient's and physician's global assessment, BASDAI and ASDAS were significantly lower in adalimumab compared with placebo at week 6 as well as at week 12 (figure 1).
Mean changes in disease activity from baseline to week 12 by treatment group
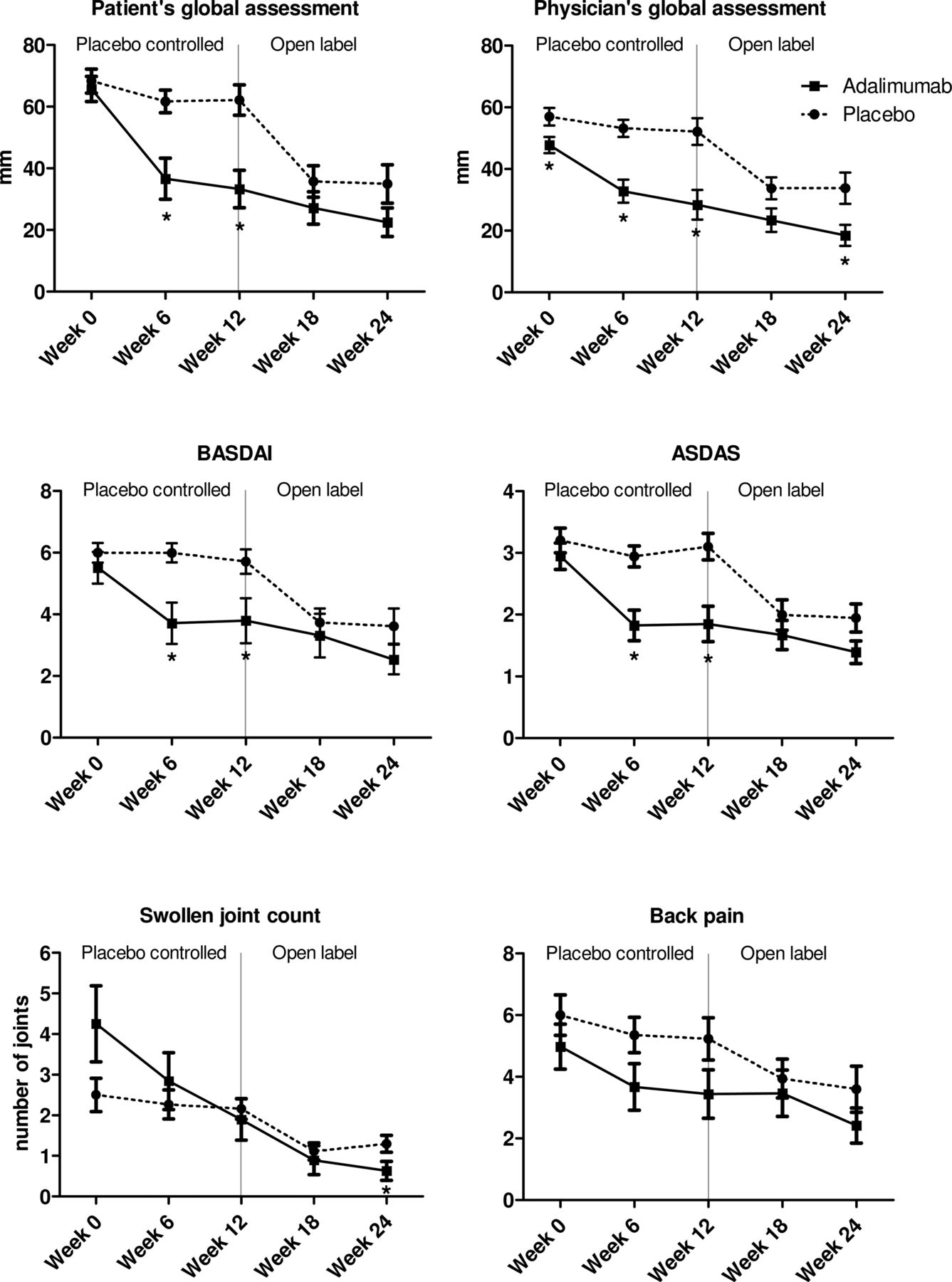
Changes in the clinical disease activity parameters during treatment with adalimumab or placebo from week 0 until 12, and during the open label extension phase with adalimumab from week 12 until 24. The panels represent the patient's and physician's global assessment of disease activity on a 100 mm visual analogue scale, the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), Ankylosing Spondylitis Disease Activity Score (ASDAS), 66 swollen joint count and back pain score (assessed by BASDAI question #2). Data are presented as mean (SEM). *p Value <0.05 compared with placebo by an independent sample t test.
Moreover, the intragroup analysis demonstrated a strong and significant improvement at week 12 compared with baseline in the adalimumab group for the patient's and physician's global assessment, SJC66, BASDAI and ASDAS (p<0.05 for all parameters) (table 3). In sharp contrast, none of these parameters showed any statistically significant change in the placebo group over this time period. The inflammatory parameters showed a numerical but non-significant decrease at week 12 of adalimumab treatment. Subanalysis of patients with increased CRP at baseline, however, showed that CRP levels decreased in seven out of seven patients treated with adalimumab.
Disease activity parameters at weeks 0, 12 and 24 by treatment group
Improvement of disease activity at week 24
After the 12-week placebo-controlled phase all patients were treated in an open label phase with adalimumab for an additional 12 weeks. In the original placebo group, adalimumab treatment induced a significant decrease of all clinical disease activity parameters between weeks 12 and 24 (p<0.05 for all parameters) (table 3 and figure 1). Additionally, ESR was significantly suppressed (p<0.05). Analysis of patients with increased CRP at baseline showed a decrease in four out of five patients during this treatment period.
In the patients who already received adalimumab from baseline, we observed a further improvement of all clinical disease activity parameters between weeks 12 and 24, reaching statistical significance for physician's global assessment, SJC66 and BASDAI (table 3 and figure 1). All clinical disease activity parameters as well as ESR were significantly improved at week 24 in comparison with baseline.
ASDAS and BASDAI response criteria
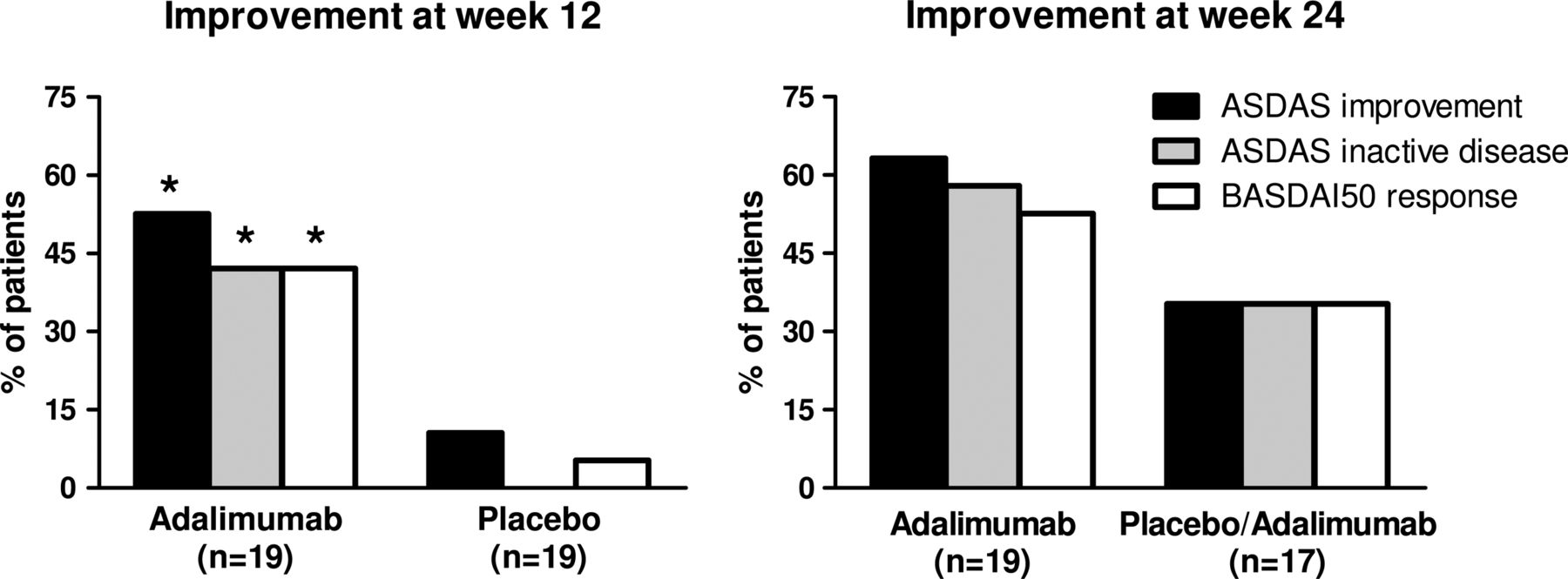
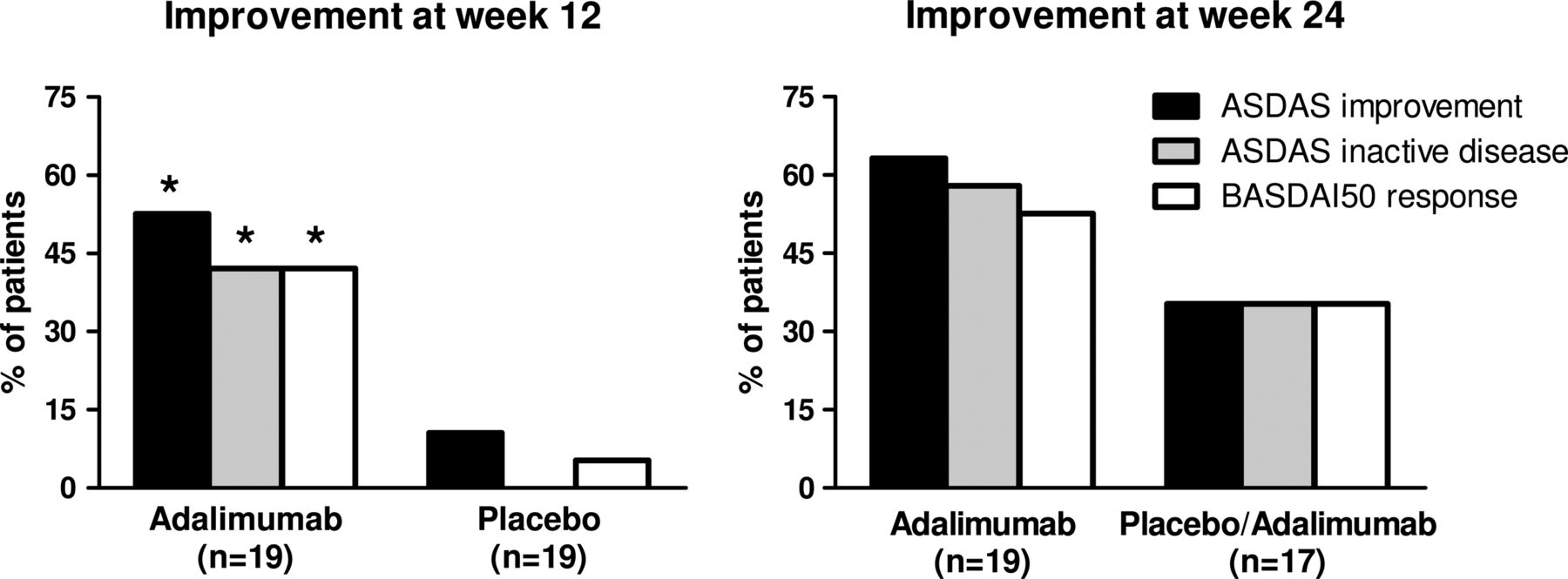
Although the trial was designed to assess the effect of adalimumab on peripheral SpA, 22 out of 40 patients had also axial complaints at baseline. As we observed a significant effect on BASDAI and ASDAS scores (tables 2, 3 and figure 1), we additionally assessed the ASDAS and BASDAI response criteria. At week 12, 53% of the patients treated with adalimumab met the criteria for ASDAS clinically important improvement, 42% the criteria for ASDAS inactive disease and 42% a BASDAI50 response (figure 2). All three assessments reached statistical significance compared with placebo, which showed an ASDAS improvement of 11%, ASDAS inactive disease of 0% and BASDAI50 response of 5%.
By 24 weeks, the initial placebo treated group showed a similar response to 12 weeks of adalimumab treatment: ASDAS clinically important improvement, ASDAS inactive disease and BASDAI50 response were reached in 35% for all three parameters (figure 2). In the group receiving adalimumab from the beginning, treatment with adalimumab for an additional 12 weeks even further increased the percentage of patients who reached the ASDAS clinically important improvement, ASDAS inactive disease and BASDAI50 response to 63%, 58% and 53%, respectively (figure 2).
{kind=link}
{kind=link}
Percentages of patients who achieved a clinically important improvement and inactive disease according to the Ankylosing Spondylitis Disease Activity Score (ASDAS) criteria and Bath Ankylosing Spondylitis Disease Activity Index 50 (BASDAI50) response at weeks 12 and 24. The first 12 weeks patients received either adalimumab or placebo, and the last 12 weeks all the patients received adalimumab in the open label extension phase. *p Value <0.05 compared with placebo by χ2 test.
Improvement in disability and QoL measurements
To assess the impact of adalimumab treatment on disability and QoL, we measured the HAQ-DI and the HUI-3. As shown in table 3, an improvement in HAQ-DI was observed at week 12 in the adalimumab group but not the placebo group. A similar trend was seen for the HUI-3. At week 24, both parameters were significantly improved in both treatment arms (p<0.05 for both parameters).
Dropouts and safety analysis
The safety analysis is summarised in table 4. In each treatment arm, 19 patients completed the 12-week placebo-controlled phase. Two patients discontinued the study because of a serious adverse event (SAE): one death due to suicide in the adalimumab group and one arthroscopy related septic arthritis in the placebo group. No other SAEs were observed. The number of non-serious adverse events (AEs) was similar (n=12 in each group). The number of infectious events was lower in the adalimumab group (n=4) than in the placebo group (n=8). Two adalimumab treated patients developed a diffuse skin rash, which resolved after topical corticosteroids and oral antihistaminic treatment, and did not result in treatment discontinuation.
AEs through week 24 by treatment group
In the open label phase, all 19 patients from the group originally treated with adalimumab completed the full study. In the group initially randomised to receive placebo, 17 patients completed the open extension until week 24: one patient dropped out due to a SAE (hospital admission because of an acute psychosis) and one patient was lost to follow-up. There were no other SAEs. Non-serious AEs included 13 infections, consisting mainly of upper respiratory tract infections and cystitis, and one local injection reaction, which resolved spontaneously. Overall, there were no unexpected safety signals.
Discussion
The present randomised double-blind, placebo-controlled clinical trial demonstrated the efficacy and safety of adalimumab in patients with active peripheral SpA not fulfilling the criteria for AS or PsA. The primary objective of the study was met as there was a rapid and significant decrease in the patient's global assessment upon adalimumab but not placebo treatment. Additionally, treatment with adalimumab but not with placebo, significantly improved all other clinical parameters of disease activity except the TJC68, which showed a numerical but not significant decrease. The efficacy of adalimumab in this patient group was confirmed by three additional observations. First, ESR and CRP as objective measurements of inflammation did consistently decrease in those patients with elevated values at baseline. Second, the patients who were initially treated with placebo also showed good efficacy upon treatment with adalimumab in the open label phase. And third, the clinical benefit of TNF-blockade was sustained and even augmented on weeks 18 and 24 in the group continuously treated with adalimumab. Moreover, multiple regression analysis (data not shown) showed that features such as gender, HLA-B27 status, CRP, ESR, SJC66 and concomitant DMARD treatment did not act as confounders in this trial.
These data are in agreement with the reported efficacy of adalimumab in large RCTs in AS42 and PsA,24 as well as in smaller trials in other SpA subtypes.1 ,4 ,11–13 27–32 Taken together, these trials support the concept that anti-TNF-therapy is highly effective in SpA as a whole independent of the phenotypic manifestation. This concept is further strengthened by the significant improvement of measurements primarily developed for axial disease such as BASDAI and ASDAS despite the fact that the trial was designed for peripheral SpA. The efficacy of adalimumab on these parameters in our study is similar to that reported for AS in the ATLAS trial: a BASDAI50 response at week 12 was achieved in 42% of our study population compared with 45% reached in the Adalimumab Trial Evaluating Long-term Efficacy and Safety for Ankylosing Spondylitis (ATLAS) trial,42 whereas at week 24, ASDAS inactive disease was reached in respectively 58% and 55% of the patients.43
The results of our trial raise three additional issues with regard to peripheral SpA. First, we used the ESSG and Amor criteria as the ASAS criteria for peripheral SpA were not published yet when the study was started. Post hoc application of these criteria to our study population, however, indicated that 38 out of the 40 patients also fulfilled the ASAS criteria for peripheral SpA. Albeit presenting primarily with peripheral disease, 22 out of 40 patients also had inflammatory back pain when actively questioned for axial complaints. Whereas strict application of the ASAS criteria would thus imply that the criteria for axial SpA should be used rather than peripheral SpA, the overlap in signs and symptoms between different SpA phenotypes observed in the present study fits with the significant improvement in BASDAI and ASDAS in these patients with predominantly peripheral SpA, and indicates that the subclassification in axial versus peripheral SpA may not always completely cover the clinical picture. It remains to be determined whether global measurements such as patient's and physician's global assessment,1 ,4 peripheral measurements such as SJC, or primarily axial measurements such as BASDAI and ASDAS are the most appropriate to monitor disease activity in SpA patients with a mixed phenotype.
A second issue is whether the improvement in disease activity also translates in improved function and QoL in non-AS, non-PsA peripheral SpA. Functional outcome parameters have been developed and validated for AS and are also applied to axial SpA but similar parameters are not available for peripheral SpA. Interestingly, also at the functional level we noticed a significant improvement of axial outcome parameters such as the lumbar mobility measured by the modified Schober index in these patients presenting primarily with peripheral disease (data not shown). As to disability and QoL, both the HAQ-DI and the HUI-3 were significantly improved at week 24. This has medical and socio-economic implications as previous studies in AS have indicated that QoL scores are associated with working status and thus with economic and societal costs.44
A third issue is that the peripheral SpA patients included in the study presented with arthritis rather than dactylitis or enthesitis (table 1). The low frequency of dactylitis, with only three patients having active dactylitis at baseline, may be related to the fact that we excluded patients fulfilling the ClASsification criteria for Psoriatic ARthritis (CASPAR) criteria for PsA. In contrast, enthesitis was noted in the history of 75% of the patients. However, only 25% of the patients had active enthesitis at inclusion, thereby precluding a detailed analysis of this manifestation in our trial. However, a previous study including specifically SpA patients with refractory heel enthesitis has indicated the efficacy of TNF-blockade by etanercept for this peripheral SpA manifestation.45
The number of SAEs, AEs and infections was similar in the adalimumab treated patients compared with the placebo treated patients. The number of SAEs was quite high in our cohort (three out of 40 patients) but two out of three SAEs (suicide and acute psychosis) were not considered to be related to the study drug. The third SAE, a septic arthritis, was most probably triggered by needle arthroscopy and occurred in the placebo group and not the adalimumab group. Although safety data remain difficult to interpret correctly in relatively small proof-of-concept trials, we did not observe unexpected safety signals.
In conclusion, the results of this randomised double-blind, placebo-controlled clinical trial indicate that SpA patients with peripheral disease who do not fulfil the criteria for AS or PsA may benefit from adalimumab treatment in terms of disease activity and QoL. This warrants further confirmation in larger studies.
Acknowledgments
We thank Abbott for the supply of the study medication for this investigator initiated and independent study. Also we would like to thank the following rheumatologists for referring patients: Dr ZN Jahangier, Dr G Collee, Dr MHW de Bois and Drs A van Sijl. We thank AN Scholten, A van Tillo, Dr DM Gerlag, R Heinrich, A Giesbers and L Gomes for their help in this study, and Professor Dr R Landewé for critical reading of the manuscript. Professor Dr DL Baeten is supported by a Vidi grant from The Netherlands Organization for Scientific Research (NWO).
References
Footnotes
Handling editor Tore K Kvien
-
Contributors JEP participated in the execution of the trial, analysis and interpretation of the data, and wrote the manuscript. LDR contributed to the design of the trial and the execution of the trial. TFH and CAA participated in the execution of the trial. KV, HJD and PPT reviewed the manuscript for content. DLB contributed to the design of the trial, interpretation of the data and supervised the development of the manuscript.
-
Competing interests Professor PP Tak is currently an employee of GlaxoSmithKline.
-
Ethics approval This study was conducted with the approval of the ethics committee of the Academic Medical Center/University of Amsterdam, The Netherlands.
-
Funding None.
-
Patient consent All patients gave written informed consent.
-
Data sharing statement Data of our research article are available upon request. Additional data of this cohort are expected to be published in the future.
-
Provenance and peer review Not commissioned; externally peer reviewed.