Article Text

Abstract
Objectives To compare the pharmacokinetics (PK), safety and efficacy of innovator infliximab (INX) and CT-P13, a biosimilar to INX, in patients with active ankylosing spondylitis (AS).
Methods Phase 1 randomised, double-blind, multicentre, multinational, parallel-group study. Patients were randomised to receive 5 mg/kg of CT-P13 (n=125) or INX (n=125). Primary endpoints were area under the concentration-time curve (AUC) at steady state and observed maximum steady state serum concentration (Cmax,ss) between weeks 22 and 30. Additional PK, efficacy endpoints, including 20% and 40% improvement response according to Assessment in Ankylosing Spondylitis International Working Group criteria (ASAS20 and ASAS40), and safety outcomes were also assessed.
Results Geometric mean AUC was 32 765.8 μgh/ml for CT-P13 and 31 359.3 μgh/ml for INX. Geometric mean Cmax,ss was 147.0 μg/ml for CT-P13 and 144.8 μg/ml for INX. The ratio of geometric means was 104.5% (90% CI 94% to 116%) for AUC and 101.5% (90% CI 95% to 109%) for Cmax,ss. ASAS20 and ASAS40 responses at week 30 were 70.5% and 51.8% for CT-P13 and 72.4% and 47.4% for INX, respectively. In the CT-P13 and INX groups more than one adverse event occurred in 64.8% and 63.9% of patients, infusion reactions occurred in 3.9% and 4.9%, active tuberculosis occurred in 1.6% and 0.8%, and 27.4% and 22.5% of patients tested positive for anti-drug antibodies, respectively.
Conclusions The PK profiles of CT-P13 and INX were equivalent in patients with active AS. CT-P13 was well tolerated, with an efficacy and safety profile comparable to that of INX up to week 30.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Innovator infliximab (INX), a chimeric monoclonal antibody (mAb) to tumour necrosis factor-α (TNFα), was the first TNF antagonist shown to be efficacious in ankylosing spondylitis (AS).1 INX significantly improved the signs, symptoms, functional status, and quality of life (QOL) of patients with AS in clinical trials, with clinical improvement seen as early as 2 weeks after initiation of therapy and an acceptable safety profile.2–4 In the Ankylosing Spondylitis Study for the Evaluation of Recombinant Infliximab Therapy (ASSERT) trial, patients receiving INX also showed significant improvement versus placebo in 20% and 40% improvement response according to Assessment in Ankylosing Spondylitis International Working Group criteria (ASAS20/ASAS40), Bath Ankylosing Spondylitis Disease Activity Index (BASDAI), Bath Ankylosing Spondylitis Functional Index (BASFI) and Bath Ankylosing Spondylitis Metrology Index (BASMI), chest expansion and physical component summary score of the SF-36.2 INX and other anti-TNF agents have become important components of the management of patients with active AS.5 ,6 With current biologic therapies approaching patent expiration, there has been considerable interest in developing biosimilar products, which are highly similar but not identical and not ‘bioidentical’, to approved ‘reference’ agents.7
CT-P13 is an IgG1 chimeric human-murine mAb biosimilar to INX. CT-P13 is produced in the same type of cell-line (Sp2/0-AG14—purchased from ATCC, Cat. CRL-1581) and has an identical amino acid sequence to INX. CT-P13 and INX have demonstrated comparable in vitro primary pharmacodynamics (PD) in a range of studies (CELLTRION, Inc. Unpublished data (see online supplementary appendix A)). CT-P13 and INX showed comparable binding affinities to monomeric and trimeric forms of human TNFα (hTNFα), transgenic mouse hTNFα (tmhTNFα) expressed by Jurkat cells and to Fcγ receptors and FcRn. Comparable hTNFα neutralising activity against a TNFα-sensitive mouse sarcoma cell-line (WEHI-164) has also been demonstrated. CT-P13 and INX are also comparable in terms of: lack of binding activity to human TNFβ and TNFα from a range of different species known not to bind infliximab; relative binding affinities to complement protein C1q; and complement-dependent cytotoxicity effects and apoptotic effects against a Jurkat T-cell-line expressing tmhTNFα. Comparable cytotoxic activities have been achieved as a result of antibody-dependent cellular cytotoxicity evaluation of human peripheral blood mononuclear cells against tmhTNF α-Jurkat T cells, demonstrating biosimilarity of CT-P13 and INX. Highly comparable human tissue cross-reactivity results have been observed for biotinylated CT-P13 and INX.
According to biosimilar guidelines from European Medicines Agency (EMA) and US Food and Drug Administration (FDA), comparative clinical trials for pharmacokinetics (PK) and efficacy are required for demonstration of clinical comparability, preferably double-blind, normally equivalence trials. Programme evaLuating the Autoimmune disease iNvEstigational drug cT-p13 in AS patients (PLANETAS) was conducted with the approval of the regulatory authorities, including the EMA. PLANETAS was not a conventional dose finding Phase 1 clinical trial but a Phase 1 biosimilar study designed to demonstrate PK equivalence and efficacy and safety comparability of CT-P13 and INX in active AS patients. Efficacy equivalence of CT-P13 and INX in a phase 3 study named Programme evaLuating the Autoimmune disease iNvEstigational drug cT-p13 in rheumatoid arthritis (RA) patients (PLANETRA).8 PK and PD endpoints were also assessed, as the indications for the PLANETAS and PLANETRA trials were different.
Patients and methods
Patients
Patients with active AS according to the 1984 modified New York classification criteria for ≥3 months prior to screening, with BASDAI score of ≥4 (range 0–10) and a visual analogue scale score for spinal pain of ≥4 (range 0–10) were eligible for PLANETAS study. Patients were permitted to receive both oral glucocorticoids (equivalent to ≤10mg daily prednisolone) and nonsteroidal anti-inflammatory drugs, if they had received a stable dose for ≥4 weeks prior to screening. Additional details of patient eligibility criteria are provided online (see online supplementary appendix B).
Study design
This study (ClinicalTrials.gov NCT01220518) was conducted according to the Declaration of Helsinki and International Committee on Harmonisation good clinical practices. The protocol was reviewed and approved by regulatory authorities and the ethics committees of each study site. Written informed consent was obtained from all patients. The study was conducted at 46 sites across 10 countries in Europe, Asia and Latin America.
Patients were randomly assigned 1:1 to receive either 5 mg/kg of CT-P13 (CELLTRION INC, Incheon, Republic of Korea) or INX (Janssen Biotech Inc, Horsham, Pennsylvania, USA), both administered by 2-h IV infusion, at weeks 0, 2, 6 and then q8 weeks up to week 30. Patients were premedicated with antihistamine (chlorpheniramine 2–4 mg or dose of equivalent antihistamine, eg, 10 mg of cetirizine) 30–60 min prior to the start of infusion at the investigator's discretion.
Patients underwent clinical assessments and blood sampling at baseline, weeks 14 and 30. At each visit, patients were questioned about adverse events (AEs) and concomitant medications and were monitored for any clinical signs and symptoms of tuberculosis (TB). Additional study details are provided in (see online supplementary appendix C).
Study endpoints
The primary endpoint was to demonstrate PK equivalence at steady state (area under the concentration-time curve (AUC] and observed maximum serum concentration (Cmax,ss)) between CT-P13 and INX assessed between weeks 22 and 30 (doses 5 and 6). Serum blood samples for PK analysis were obtained immediately prior to the study treatment infusion, at the end of the infusion and 1 h after the study treatment infusion. For primary PK analysis, a total of 10 serum blood samples were obtained between weeks 22 and 30. All PK analyses were conducted using a flow-through immunoassay platform (GyrolabxP; Gyros AB, Sweden).
In an equivalence trial, we conclude that two treatments are equivalent if the observable difference (ΔE) between them lies within an established interval for predefined clinical equivalence margin (−d, d). In general, the ‘null hypothesis’ is that the difference (ΔE) is outside of the equivalence margin, that is, either ΔE>d or ΔE<–d. If collected data on the true difference ΔE reject the null hypothesis of ‘non-equivalence’ then we can accept the alternative explanation (−d≤ΔE≤d) that the two treatments work equally well.9 An equivalence margin of 80–125% was selected based on recommendations for bioequivalence trials.10–13 The use of 90% confidence intervals (CIs), lying within the equivalence margin of 80–125%, was therefore considered to be the best available method of determining bioequivalence for PK comparative trials. In our PK analysis, the predetermined difference is defined by its corresponding ratio since we use the ratio of geometric means of PK endpoints.
Secondary endpoints included additional PK, efficacy, immunogenicity and safety parameters. The secondary PK endpoints included assessments of observed maximum serum concentration (Cmax), minimum serum concentration (Cmin), time to reach Cmax (Tmax) up to week 30 and the comparison of the following parameters from week 22 to 30: average concentration at steady state (Cav,ss); minimum concentration at steady state immediately before the next infusion (Cmin,ss); swing ((Cmax,ss−Cmin,ss)/Cmin,ss); degree of fluctuation ((Cmax,ss−Cmin,ss)/Cav,ss); mean residence time (MRT); terminal elimination half-life calculated between doses 5 and 6 (T1/2); total body clearance (CLss) and volume of distribution at steady state (Vss).
Efficacy endpoints were assessed at weeks 14 and 30 and included: proportion of patients achieving ASAS20 or ASAS40 responses; Ankylosing Spondylitis Disease Activity Score (ASDAS) score; change in BASDAI, BASFI and BASMI scores versus baseline; change in chest expansion score versus baseline; and QOL (assessed using the Medical Outcomes Study Short-Form Health Survey (SF-36)).
Blood samples were assessed for anti-drug antibodies (ADA) at weeks 14 and 30. Antibodies against CT-P13 or INX were measured using an electrochemiluminescent immunoassay method utilising the Meso Scale Discovery platform (MSD, Rockville, Maryland, USA).
Safety endpoints included incidence and type of AEs, serious AEs (SAEs) and incidence of infusion-related reactions, infection and changes in clinical laboratory parameters from baseline. AEs were coded using the Medical Dictionary for Regulatory Activities and severity was characterised as mild, moderate or severe. ECGs were recorded at week 30.
All patients were screened for latent or active TB by an interferon gamma release assay (IGRA) utilising QuantiFERON-TB Gold in tube (Cellestis Ltd, Australia) and chest x-ray. Patients with latent TB received prophylactic medication according to the local guidelines. For countries with an increased incidence of TB, IGRA was used to identify positive conversion from negative results at weeks 14 and 30, in line with the WHO recommendations for sole use of IGRA in non-HIV adults receiving anti-TNF therapy.14 ,15
Statistical analysis
Sample size was determined using the following criteria: a coefficient of variation (CV) of 50%, expected ratio of means=1, 2-sided α=0.1, power=90%, and a 2-sided equivalence margin of 80–125% for AUC and Cmax,ss.16 ,17 Recruitment of 196 patients were required to demonstrate an effect. Allowing for a drop-out rate of 20%, a minimum of 246 patients were required for randomisation.
The PK population consisted of all patients who received at least the first five doses of study treatment and provided an end of infusion sample and at least one post-treatment PK sample to facilitate calculation of AUC and Cmax,ss. The PK population included only patients who did not have any major protocol deviations (see online supplementary appendix D).
Primary endpoints were assessed by statistical comparison of AUC and Cmax,ss. Serum concentrations and PK parameters were summarised using quantitative descriptive statistics (including geometric mean and CV) by actual treatment group and study visit (and by time point for serum concentrations).
Efficacy analysis assessed the proportion of patients achieving clinical response (ASAS20/ASAS40) by logistic regression modelling including all randomised patients, with treatment as a fixed effect and the stratification factors (region, baseline BASDAI score) as covariates. Treatment effect was estimated by calculating the OR and 95% CI. Descriptive statistics for actual result and change from baseline were calculated for the following quantitative parameters: ASDAS, BASDAI, BASFI, BASMI, chest expansion and SF-36.
The safety population consisted of all patients who received at least one (full or partial) dose of either of the study treatments during any dosing period. In this population, patients were included in the CT-P13 group for safety analyses irrespective of their randomisation if they received at least one (full or partial) dose of CT-P13.
Safety analysis was performed by presenting data on hypersensitivity, ECG results, physical examination, vital sign measurements, clinical laboratory tests (haematology, clinical chemistry and urinalysis), AEs, concomitant medications and immunogenicity.
Results
Patients
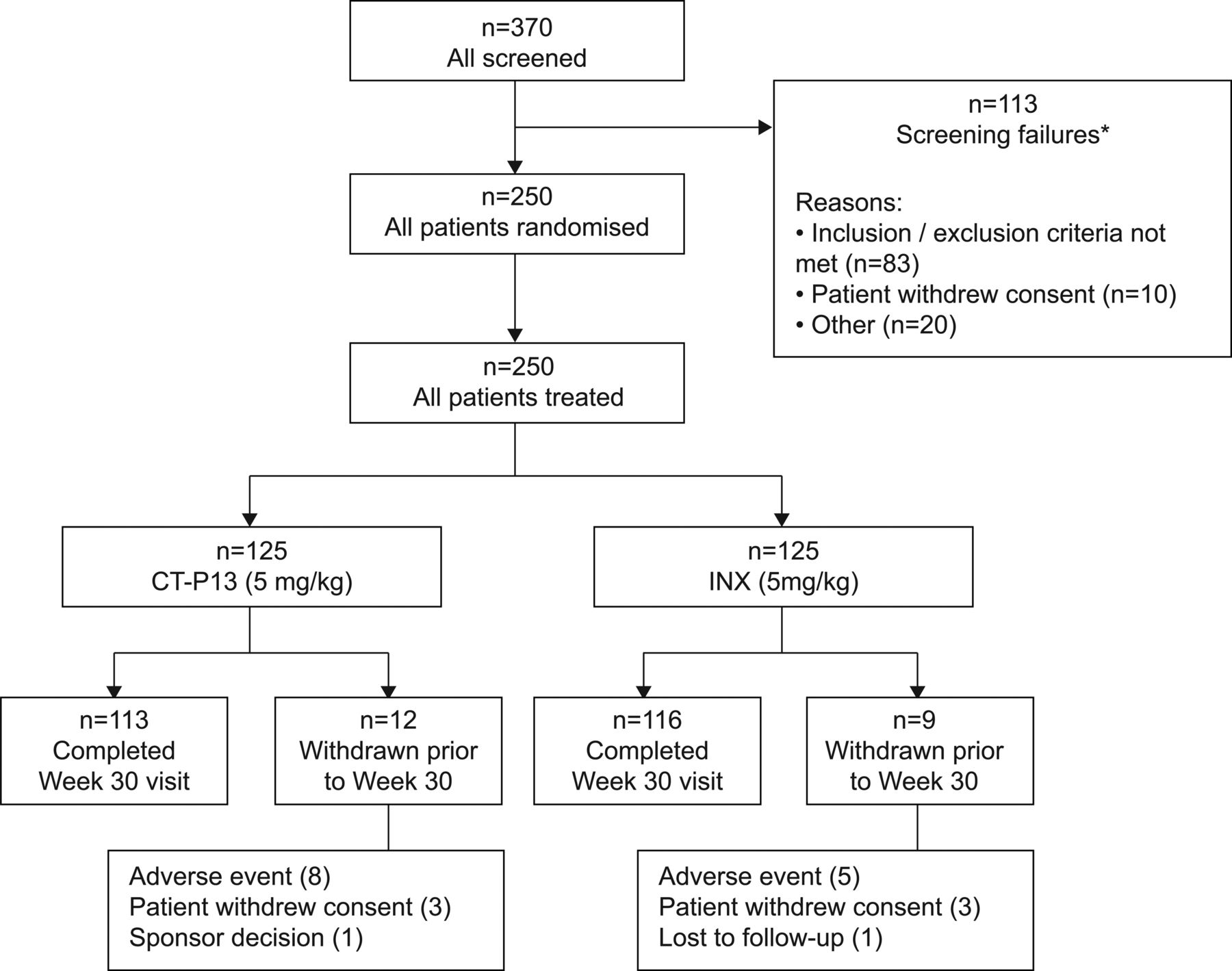
The first patient was screened in November 2010; the week 30 evaluation of the last patient was performed in December 2011. Baseline demographics are shown in table 1. Of the 250 randomised patients, 229 completed the 30-week study period and 21 discontinued study treatment prior to week 30, primarily due to AEs (5.2%) and patient withdrawal of consent (2.4%) (figure 1).
Baseline demographics*

Flowchart of patient disposition. A total of 370 patients were screened for the study, and 250 eligible patients were randomised to the CT-P13 group (N=125), and the innovator infliximab (INX) group (N=125) to receive 5 mg/kg of CT-P13 or INX, respectively. All 250 randomly assigned patients were included in the intent-to-treat population. *Seven patients (three from CT-P13 group, four from INX group) from a potentially fraudulent study site were excluded from analyses.
PK analyses included 223 patients. Efficacy and safety analysis were performed in all 250 patients.
Pharmacokinetics
Steady state PK (AUC and Cmax) was equivalent for CT-P13 (32765.8 μgh/ml and 147.0 μg/ml) and INX (31359.3 μgh/ml and 144.8 μg/ml) in the overall PK population (table 2 and figure 2). The ratio of geometric means was near 100% for AUC and Cmax,ss. In the ADA-negative subset of patients (n=171), geometric means of AUC and Cmax,ss were higher than in the overall PK population, but the ratios of geometric means in this subgroup remained near 100% for both measures. The mean secondary PK endpoints—Cav,ss, Cmin,ss, swing, degree of fluctuation, MRT, T1/2, CLss, Vss, Cmax, Cmin and Tmax—were also highly similar between CT-P13 and INX (table 3).
Overall steady state PK between weeks 22 and 30
Mean (CV) Serum pharmacokinetic parameters of INX: pharmacokinetic population

{kind=link}
{kind=link}
Mean (±SD) serum concentrations of innovator infliximab (INX) and CT-P13 versus time by treatment. Serum concentration of drug was measured using a flow-through immunoassay platform (GyrolabxP). Mean serum drug concentration profiles of CT-P13 and INX were plotted by treatment on scheduled sample times. (A) Mean serum drug concentration following administration of Dose 5 (10 scheduled sample times between weeks 22 and 30) of CT-P13 (5 mg/kg) or INX (5 mg/kg). (B) Mean serum drug concentration of CTP13 and INX following administration of Doses 1–6. Blood samples were obtained 15 min prior to infusion, at the end of the infusion and 1 h postinfusion.
Clinical efficacy
Efficacy was highly similar between the two groups, as measured by all efficacy endpoints. ASAS20 response was achieved in 62.6% and 70.5% for CT-P13 and 64.8% and 72.4% for INX at weeks 14 (OR=0.91, 95% CI 0.53 to 1.54) and 30 (OR=0.91, 95% CI 0.51 to 1.62), respectively. ASAS40 response was achieved in 41.7% and 51.8% for CT-P13 and 45.9% and 47.4% for INX at weeks 14 (OR=0.85, 95% CI 0.51 to 1.42) and 30 (OR=1.19, 95% CI 0.70 to 2.00), respectively (see online supplementary appendix E).
The mean change from baseline in the ASDAS-CRP score was highly similar for both treatment groups at weeks 14 (−1.8; SD=1.1 vs −1.8; SD=1.1) and 30 (−1.8; SD=1.2 vs −1.7; SD=1.2) for CT-P13 and INX, respectively.
The median change from baseline to weeks 14 and 30 for CT-P13 and INX was noted in the following secondary endpoints: BASDAI score (week 14: −2.7 vs −2.7 and 30: −3.1 vs −2.5); BASFI score (week 14: −2.2 vs −2.4 and 30: −2.6 vs −2.2); BASMI score (week 14:0.0 vs 0.0 and 30: −1.0 vs −1.0); and chest expansion score (week 14: +0.2 vs +0.5 and 30: +0.5 vs +0.5), respectively. Similarly, SF-36 score increased from baseline to week 14 and 30 similarly at both time points and in both treatment groups. At week 30, a significant improvement from baseline in the physical component score of the SF-36 (median change from baseline 7.6 vs 8.5, respectively) was observed in both treatment groups. A similar effect was observed for the mental component score of the SF-36 (6.5 vs 5.2).
An ASAS20 response for CT-P13 and INX at week 30, assessed according to baseline C-reactive protein (CRP) levels, was achieved in 75.2% and 77.6% of patients with a baseline CRP of >3 times the upper limit of normal (ULN) (OR=0.76: 95% CI 0.32 to 1.84) and in 68.9% and 67.2% of patients with a baseline CRP ≤3×ULN, respectively (OR=0.99: 95% CI 0.45 to 2.17). The median change from baseline to week 30 in CRP and erythrocyte sedimentation rate was −0.7 mg/dl and −21.0 mm/h for CT-P13 and −0.8 mg/dl and −19.5 mm/h for INX, respectively.
Overall, no statistical significance in clinical response between the treatment groups at weeks 14 and 30 was found.
Immunogenicity
Antibodies to infliximab with active AS patients were detected in 9.1% (n=11) and 11.0% (n=13) of patients for CT-P13 and INX at week 14 and 27.4% (n=32) and 22.5% (n=25) of patients for CT-P13 and INX, respectively, at week 30.
The efficacy results were analysed for ADA-positive and ADA-negative patients in a post-hoc analysis, and it was found that ADA-positive patients had a less robust ASAS20 response (see online supplementary appendix F). No statistically significant difference between the CT-P13 and INX treatment groups was observed at week 14 and 30.
Safety
Overall treatment-emergent AEs (TEAEs) were reported in 83 (64.8%) patients and 78 (63.9%) patients from the CT-P13 and INX treatment arms, respectively (table 4). The majority of TEAEs was mild-to-moderate in intensity. The TEAEs considered by the investigator to be related to the study treatment and most frequently reported for patients were: CT-P13: increased alanine transaminase (ALT) (n=14, 10.9%) and aspartate transaminase (AST) (n=12, 9.4%), latent TB (n=5, 3.9%), urinary tract infection (n=5, 3.9%), serum creatine phosphokinase elevation (n=4, 3.1%) and γ-glutamyltransferase elevation (n=4, 3.1%); INX: increased ALT (n=13, 10.7%) and AST (n=10, 8.2%), γ-glutamyltransferase elevation (n=5, 4.1%) and latent TB (n=4, 3.3%). Infusion-related reactions occurred in five (3.9%) and six (4.9%) patients in CT-P13 and INX groups, respectively. The proportions of infusion-related reactions in CT-P13 and INX groups were 3.1% (n=1) vs 11.1% (n=3) for ADA-positive group and 3.4% (n=3) and 2.2% (n=2) for ADA-negative group, respectively.
Related treatment-emergent adverse events reported in at least 1% of patients in either treatment group, no (%)
Similar rates of SAEs were reported between treatment groups, regardless of the relationship with the study drug (see online supplementary appendix G). No deaths were reported during the study.
Discussion
This randomised, double-blind, multicentre and multinational, parallel-group, prospective PLANETAS study assessed the PK equivalence and safety and efficacy comparability of multiple doses of CT-P13 (5 mg/kg) versus INX (5 mg/kg) administered up to week 30 in active AS patients.
The primary outcome, steady-state PK (AUC and Cmax,ss), was shown to be equivalent for CT-P13 and INX (90% CIs for the mean AUC and Cmax,ss were 94–116% and 95–109%, respectively). These values were within the predefined margins for equivalence (80–125%), thereby satisfying the criteria set for PK equivalence of CT-P13 to INX. This predefined margin is considered appropriate from a clinical perspective because of the broad therapeutic window and high variability of INX.18 ,19 AUC and Cmax,ss were higher in the ADA-negative subset of patients in this study, versus values for the overall PK population. Published PK data for the 5mg/kg dose of INX in AS is sparse,20 but the values for AUC and Cmax,ss reported in this study are similar to those reported in previous studies of INX monotherapy using a similar dosing pattern in Crohn's disease.21 The coadministration of methotrexate (MTX) is thought to increase concentrations of INX in patients with rheumatoid arthritis (RA).22–25 Although coadministration of MTX with INX in AS is not recommended,5 ,6 the effect of coadministration on CT-P13 should be further studied—especially in patients with peripheral arthritis. In patients with AS, the possible influence of ADA on circulating concentrations of INX, including the potential consequence, an impaired clinical response, has been studied but yielded conflicting results.26–28 Nevertheless, INX monotherapy remains a useful treatment for the majority of patients with AS, for which long-term data are available.29 ,30
The mean secondary PK endpoints were also highly similar between CT-P13 and INX groups. CT-P13 was also equivalent to INX up to week 30 in terms of efficacy as assessed by ASAS20/ASAS40 criteria. The median change from baseline in BASDAI, BASFI, BASMI, chest expansion and SF-36 score was highly similar at weeks 14 and 30, underlining the benefit of CT-P13 and INX in both physician-measured and patient-reported outcomes.
The efficacy outcomes of this trial were comparable to those reported previously in pivotal randomised controlled trials of INX in AS.1 ,2 The ASAS20 and ASAS40 responses at week 30 in this study for INX (72.4% and 47.4%, respectively) are similar to those reported at week 24 in the ASSERT trial (61.2% and 47.0%, respectively). The magnitude of improvements in secondary efficacy endpoints with INX in this trial were also comparable to those reported in ASSERT.2 Improvements from baseline to week 30 of the physical component of the SF-36 in the INX group in this study were comparable to those reported in ASSERT (8.5 vs 10.2, respectively), as were the median baseline scores of the physical component of the SF-36 (28.8 vs 33.1, respectively).2 However, the improvements from baseline to week 30 of the mental component of the SF-36 in this study were not seen in ASSERT (5.2 vs 2.7, respectively), likely reflecting the higher baseline median scores of the mental component of SF-36 in ASSERT (37.2 vs 47.6, respectively).2 ASDAS C was assessed in this study, and values were found to be highly similar between treatment groups at both weeks 14 and 30.31 Comparison of ASAS20 and ASAS40 values for CT-P13 and INX by ADA status showed no statistical significance despite the observational difference in the ADA-positive subset, the latter probably due to the lack of patient numbers.
Overall, CT-P13 and INX were well-tolerated and their safety profiles were comparable. The majority of patients had negative immunogenicity results according to the electrochemiluminescent immunoassay method at weeks 14 and 30. Extra-articular manifestations such as uveitis, psoriasis and inflammatory bowel disease were not assessed in this study, but the single cases of uveitis and psoriasis in the INX group were reported as AEs. The incidence of AEs, SAEs, infections and infusion-related reactions with INX in this trial were comparable to that seen in ASSERT.2
There were also comparably low rates of TB up to week 30. The incidence of TB in our study (1.2% overall: CT-P13 (n=2), INX group (n=1)) is higher than in ASSERT, a trial of INX in AS, in which no cases were reported during the study period.2 A difference in the study site locations may partly explain this; ASSERT included only centres in North America and Western Europe, whereas our trial included centres in countries known to have higher TB incidence. Interestingly, the incidence of active TB in our study is similar to those of the two major trials of the 3 mg/kg dose of INX in RA, Anti-TNF Trial inRheumatoid Arthritis with Concomitant Therapy (ATTRACT) (0.3%) and Active-Controlled Study of Patients Receiving Infliximab for the Treatment of Rheumatoid Arthritis of Early Onset (ASPIRE) (0.5%), which included centres only in North America and Western Europe.18 ,32
According to the National Institute of Health and Clinical Excellence (NICE) incremental cost-effectiveness ratios (ICERs), INX has not been recommended for treatment of AS because NICE experts did not consider it to be cost-effective. However, this decision should be revisited based on the results of this study of CT-P13 in AS and its lower future price. This may have a potentially positive impact on healthcare costs.
In this PLANETAS study, PK equivalence for CT-P13 and INX was demonstrated. There was no significant difference in efficacy and safety data. Although only comparable clinical efficacy was shown in this study, clinical bioequivalence was investigated in a Phase 3 study in RA.8
The positive results of this study provide a rationale for future studies of CT-P13 in the treatment of other TNF-mediated inflammatory diseases.
Conclusions
CT-P13 and INX were shown to be equivalent in terms of AUC and Cmax,ss in patients with active AS. Clinical efficacy endpoints, including ASAS20 and ASAS40 responses, were highly similar between CT-P13 and INX groups. CT-P13 was well-tolerated with an immunogenicity and safety profile comparable to that of INX up to week 30.
Acknowledgments
The authors wish to thank the patients and study personnel who made this trial possible, and the PLANETAS study investigators: Bulgaria; Kadinov V, Rashkov R; Chile; Aguirre V, Goecke Sariego I, Gutierrez M; Colombia; Otero Escalante WJ; Korea (Republic of); Kang SW, Kim HY, Kim TH, Park YB, Park YE, Song JS, Suh CH; Latvia; Andersone D, Saulite-Kandevica D; Mexico; Abud-Mendoza C, Araiza R, Cons Molina F, Morales-Torres J, Pacheco-Tena C; Poland; Brzezicki J, Brzosko M, Jaworski J, Korkosz M, Krogulec M, Piotrowski M, Ruzga Z, Wiland P; Portugal; Vaz Patto J; Spain; Blanco Garcia F, Díaz-González F; Ukraine; Amosova K, Dyadyk O, Gnylorybov A, Smiyan S, Ter-Vartanian S; USA; Ahn C. Editorial support (writing assistance, assembling tables and figures, collating author comments, grammatical editing and referencing) was provided by Vanessa Lane, Peter Hopkins and Rory Elsome (TVF BioLogic, London, UK) and was funded by CELLTRION Inc, Incheon, Republic of Korea.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online appendix A
- Data supplement 2 - Online appendix B
- Data supplement 3 - Online appendix C
- Data supplement 4 - Online appendix D
- Data supplement 5 - Online appendix E
- Data supplement 6 - Online appendix F
- Data supplement 7 - Online appendix G
Footnotes
-
Handling editor Tore K Kvien
-
Contributors WP, SJL and HUK were involved in the conception and design of the study, acquisition of data and/or analysis and interpretation of data; drafting of manuscript and revising it critically for important intellectual content; final approval of the version to be published. DHY and JB were involved in the conception and design of the study; drafting of manuscript and revising it critically for important intellectual content; final approval of the version to be published. PH, SJ, VK, GL, PM, HM, SGU, MJL and YAL were involved in the acquisition of data; drafting of manuscript and revising it critically for important intellectual content; final approval of the version to be published. The project management, clinical and medical monitoring, pharmacovigilance (PVG), data management, analysis of pharmacokinetic (PK) data, biostatistical analysis, and medical writing were performed under contract with PPD, Inc. in collaboration with the CELLTRION, Inc.
-
Funding This study was funded by CELLTRION Inc.
-
Competing interests All authors have completed the Unified Competing Interest form at http://www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: financial support for the submitted work from Celltrion: WP, DHY, PH, VK, PM and YAL received research grants and, or consultancy fees, and logistics support; SJL, SJ, GL, HM, SGU and MJL received research grants and/ or consultancy fees only; HUK is a full-time employee of Celltrion. JB has received payment for consultancy and lectures from Celltrion.
-
Patient consent Obtained.
-
Ethics approval The protocol was reviewed and approved by each site's institutional review board or independent ethics committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Data sharing statement All data available for this paper are included in the manuscript and online supplementary appendices.
Linked Articles
- Editorial
- Clinical and epidemiological research