Article Text

Abstract
Objective The aim of this study was to assess long-term golimumab therapy in patients with rheumatoid arthritis (RA) who discontinued previous tumour necrosis factor alpha (TNFα) inhibitor(s) for any reason.
Methods Results through week 24 of this multicentre, randomised, double-blind, placebo-controlled study of active RA (≥4 tender, ≥4 swollen joints) were previously reported. Patients received placebo (Group 1), 50 mg golimumab (Group 2) or 100 mg golimumab (Group 3) subcutaneous injections every 4 weeks. Patients from Groups 1 and 2 with <20% improvement in tender/swollen joints at week 16 early escaped to golimumab 50 mg and 100 mg, respectively. At week 24, Group 1 patients crossed over to golimumab 50 mg, Group 2 continued golimumab 50/100 mg per escape status and Group 3 maintained dosing. Data through week 160 are reported.
Results 459 of the 461 randomised patients were treated; 236/459 (51%) continued treatment through week 160. From week 24 to week 100, ACR20 (≥20% improvement in American College of Rheumatology criteria) response and ≥0.25 unit HAQ (Health Assessment Questionnaire) improvement were sustained in 70–73% and 75–81% of responding patients, respectively. Overall at week 160, 63%, 67% and 57% of patients achieved ACR20 response and 59%, 65% and 64% had HAQ improvement ≥0.25 unit in Groups 1, 2 and 3, respectively. Adjusted for follow-up duration, adverse event incidences (95% CI) per 100 patient-years among patients treated with golimumab 50 mg and 100 mg were 4.70 (2.63 to 7.75) and 8.07 (6.02 to 10.58) for serious infection, 0.95 (0.20 to 2.77) and 2.04 (1.09 to 3.49) for malignancy and 0.00 (0.00 to 0.94) and 0.62 (0.17 to 1.59) for death, respectively.
Conclusion In patients with active RA who discontinued previous TNF-antagonist treatment, golimumab 50 and 100 mg injections every 4 weeks yielded sustained improvements in signs/symptoms and physical function in ∼57–67% of patients who continued treatment. Golimumab safety was consistent with other anti-TNF agents, although definitive conclusions regarding long-term safety require further monitoring.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Tumour necrosis factor alpha (TNFα) inhibitors have been used to treat rheumatoid arthritis (RA) for >10 years. Patients with insufficient response to TNF inhibitors are routinely switched to other biological agents, including other TNF inhibitors. Thus, increasingly more patients with RA have previous experience with ≥1 TNF inhibitor. Among the newer anti-TNF agents, golimumab is a human monoclonal anti-TNF agent administered subcutaneously every 4 weeks.
GO-AFTER (GOlimumab After Former antitumour necrosis factor α Therapy Evaluated in Rheumatoid arthritis) was the first prospective, randomised, phase 3, double-blind, placebo-controlled trial to assess a TNF inhibitor in patients with active RA who previously received TNF inhibitor(s). These patients had also received several disease-modifying antirheumatic drugs (DMARDs) prior to TNF inhibitor(s), thereby representing a difficult-to-treat population. Treatment with golimumab 50 mg and 100 mg every 4 weeks versus placebo yielded significantly higher ACR20 (≥20% improvement in American College of Rheumatology criteria) response rates at week 14 (35% and 38% vs 18%, respectively; both p<0.001) and no unexpected safety concerns through week 24.1 Efficacy and safety findings through week 160 of the GO-AFTER long-term extension (LTE) are reported herein.
Patients and methods
GO-AFTER (NCT00299546) was conducted according to the Declaration of Helsinki. All patients provided written informed consent, and the protocol was approved by each institution's human subjects ethical review board.
Patients
Patient enrolment began 21 February 2006; data were collected at visits conducted through LTE week 160. Eligible patients with RA2 had active disease (≥4 swollen, ≥4 tender joints); had previously received etanercept, adalimumab or infliximab for ≥8 (adalimumab, etanercept) or ≥12 (infliximab) weeks; and could have discontinued these agents for any reason (documented as lack of efficacy, intolerance, other). Additional inclusion/exclusion criteria were previously reported.1
Study design
Patients were randomised (1:1:1) to receive subcutaneous injections of placebo, golimumab 50 mg or golimumab 100 mg every 4 weeks. Stable doses of synthetic DMARDs were allowed. Patients and investigators were blinded to treatment assignment; golimumab and placebo were supplied in identical single-use vials.
Patients in the placebo and golimumab 50 mg groups with <20% improvement in both tender and swollen joint counts at week 16 early escaped (EE) to receive golimumab 50 mg or 100 mg, respectively, at week 16 and week 20. Dosing was not changed in the 100 mg group.
GO-AFTER included a LTE. From week 24 forward, patients in the placebo group crossed over to golimumab 50 mg every 4 weeks and patients in the golimumab 50 mg group continued with golimumab 50 or 100 mg every 4 weeks per EE status. The study blind was maintained during the LTE until the week 24 database lock, after which patients receiving golimumab 50 mg could escalate to 100 mg at the investigator's discretion. Golimumab doses could not be reduced through week 160.
Procedures
Clinical response through week 160 was assessed using ACR20/50/70,3 28-joint count Disease Activity Score (DAS28) response (good/moderate) and DAS28 remission (score<2.6) criteria.4,–,6 DAS28 scores were determined using erythrocyte sedimentation rate (ESR) and C reactive protein (CRP) with established cut points for disease activity states.7 Clinical remission according to ACR–EULAR (European League Against Rheumatism) criteria was also evaluated using the Simplified Disease Activity Index (SDAI score≤3.3).8 ,9 Physical function was assessed using the Health Assessment Questionnaire (HAQ).10 Adverse events (AEs) were coded according to MedDRA.1
Data analysis
Clinical outcomes through week 160 are summarised as observed data by randomised treatment groups using descriptive statistics; missing data were neither replaced nor imputed. All patients remaining in the study, including those initially treated with placebo, received golimumab for ≥2 years by week 160, precluding statistical comparisons among treatment groups. The proportions of patients achieving ACR response, DAS28 response/remission, ACR-EULAR index remission (SDAI) remission and/or ≥0.25 unit improvement in HAQ11 were determined. Changes from baseline (week 0 of main study) were also determined. Treatment groups were defined according to patients' original randomisation: (1) patients randomised to placebo, including patients who EE at week 16 or crossed over at week 24 to golimumab 50 mg and/or escalated after week 24 to 100 mg; (2) patients randomised to golimumab 50 mg, including patients who EE at week 16 or dose escalated after week 24 to 100 mg; and (3) patients randomised to golimumab 100 mg. Efficacy data from one North American site (16 patients) were excluded because of protocol violations identified during standard auditing; baseline and safety data from these patients were included. Because numbers of patients in LTE phases of double-blind trials typically decline over time, thereby yielding falsely elevated response rates among the remaining responder-enriched population,12 efficacy data are presented as (1) absolute numbers to demonstrate actual changes observed during the LTE, despite the limited numbers of patients with response data, and (2) proportions of responders based on randomised patients who continued study participation and had response data, despite the enriched nature of the resulting population.
Safety data are summarised for randomised and treated patients. Individual AEs were attributed to treatment received at onset; therefore, individual patients may appear in >1 treatment column. Standardised incidence ratios (SIRs) for malignancies were determined using the Surveillance, Epidemiology and End Results (SEER) database.13
Results
Patient disposition and characteristics
Patient recruitment began in February 2006, and data included in this report were collected through study week 160. Patient disposition through week 24 has been detailed. Overall, 57 of 461 (12.4%) randomised patients discontinued study participation through week 24, including 31/155 (20.0%), 12/153 (7.8%) and 14/153 (9.2%) patients randomised to receive placebo, golimumab 50 mg and golimumab 100 mg, respectively.1 Approximately one-half of treated patients continued study treatment through week 160 (figure 1). Common reasons for study agent discontinuation included unsatisfactory therapeutic effect (43% of discontinued patients) and AEs (28%, including nine patients with worsening RA) (figure 1). The proportions of patients discontinuing study agent due to AEs or unsatisfactory therapeutic effect increased with greater number of previous TNF antagonists (table 1).

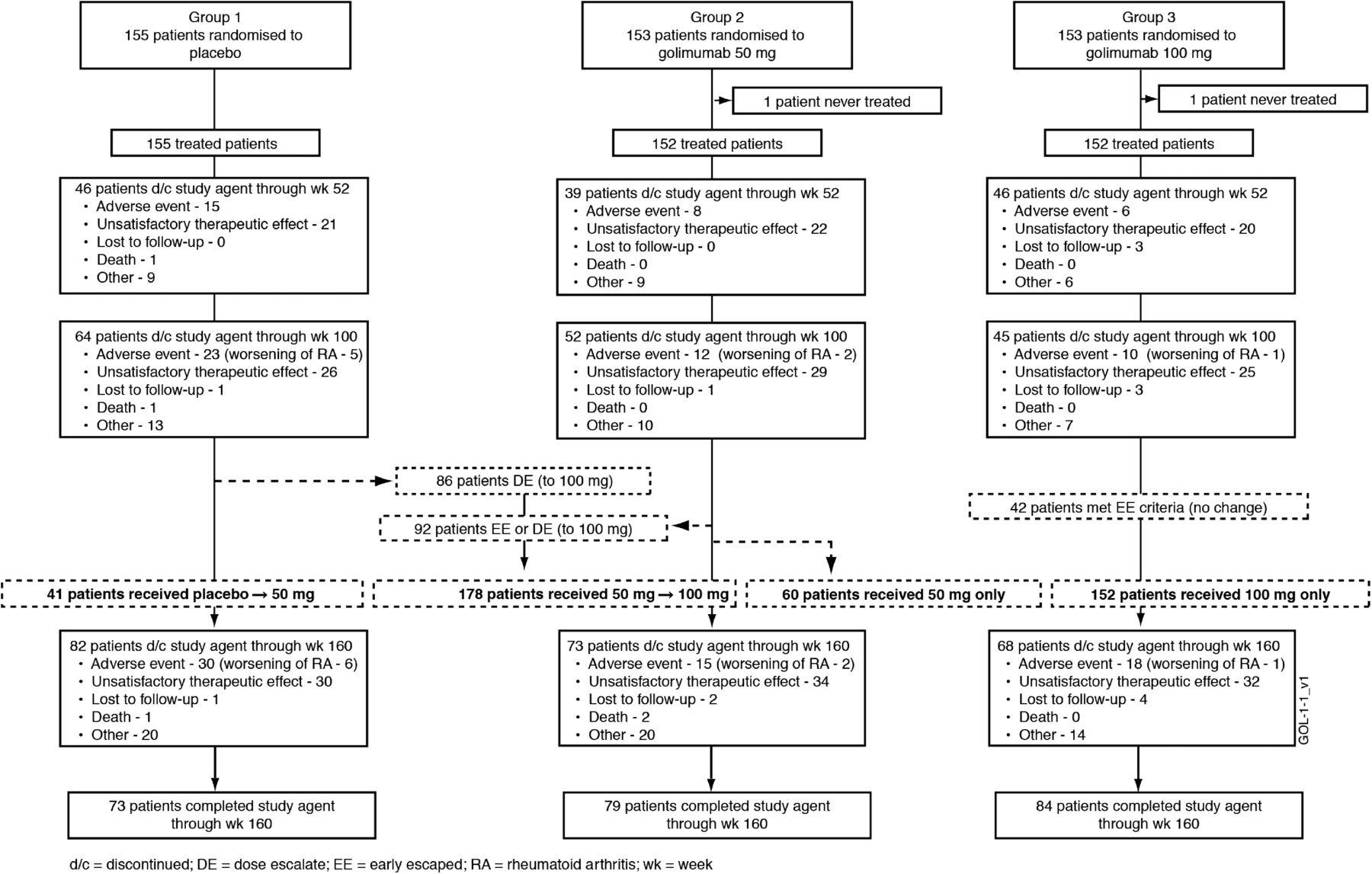{kind=link}
Patient disposition through week 160.
Summary of patient characteristics and RA medications at baseline of the GO-AFTER trial
The extent of active disease, inflammation and physical function impairment and reasons for previous TNF inhibitor(s) discontinuation were consistent across treatment groups (table 1). Overall, 178 patients dose escalated (50→100 mg) at the investigator's discretion. During the LTE, 70% (137/195) of eligible patients dose escalated (figure 1).
Clinical response
The proportions of patients achieving ACR20, DAS28-ESR response and DAS28-ESR remission at week 24 among patients who received golimumab 50 mg (34%, 46% and 10%, respectively) and 100 mg (44%, 61% and 16%, respectively) were significantly higher versus placebo-treated patients (17%, 25% and 3%, respectively; all p<0.05).1
The proportions of patients meeting the more stringent ACR50/70 response criteria appeared stable from week 52 to week 160 in all treatment groups (table 2). Similar findings were generally observed in ≥1 golimumab group(s) for DAS28-ESR response, SDAI low disease activity (>3.3–11), SDAI remission and HAQ improvement (table 2). Thus, despite smaller numbers of patients in each group over time, response rates were generally sustained.
Summary of clinical response to golimumab at weeks 52, 100 and 160 of the GO-AFTER trial
An important aspect of managing difficult-to-treat RA relates to maintenance of response. From week 24 to week 100, ACR20, DAS28 and ≥0.25 unit HAQ responses were sustained in 70–73%, 78–84% and 75–81% of responding patients, respectively (table 3).
Sustained clinical response to golimumab in the GO-AFTER trial
Among the 137 patients who dose escalated from 50 mg to 100 mg through week 160 (figure 1), within 12 weeks following dose escalation, ACR20 and ACR50 response rates increased and were generally maintained over the subsequent 88 weeks. A similar pattern was observed for DAS28 responses (table 3).
Golimumab efficacy was confirmed in patients receiving placebo→golimumab 50 mg with or without further escalation to 100 mg. Clinical and functional response rates were similar to those demonstrated by patients who received golimumab since week 0 (tables 2 and 3).
Adverse events
AEs through week 24 of GO-AFTER have been reported.1 During the double-blind, placebo-controlled phase (weeks 0–16), 70%, 61% and 73% of placebo, golimumab 50 mg and golimumab 100 mg patients, respectively, experienced AEs. Infections (28%, 27% and 25%), serious AEs (7%, 5% and 3%) and serious infections (2%, 2% and 1%) were also consistent across treatment groups. More patients receiving golimumab 100 mg (11%) had injection-site reactions versus placebo (3%) or golimumab 50 mg (4%).1
Largely because the GO-AFTER study design encompassed early escape and crossover to golimumab, average weeks of follow-up for LTE patients receiving golimumab 50 mg and 100 mg were 32 and 36 weeks at week 52, 48 and 65 weeks at week 100 and 60 and 102 weeks at week 160, respectively (table 4). Total patient-years of follow-up were 58, 319 and 645, respectively, for placebo, golimumab 50 mg and golimumab 100 mg.
Cumulative summary of golimumab safety through week 160 of the GO-AFTER trial
Through week 52, when average weeks of follow-up were comparable for 50 mg and 100 mg, 76% of patients in each group experienced ≥1 AE, indicating no dose response. By week 160, 81% (50 mg) and 90% (100 mg) of patients experienced ≥1 AE; respective incidences were 18% and 25% for serious AEs, 9% and 12% for discontinuation of study agent due to an AE and 5% and 9% for serious infections (table 4).
At week 160, the incidences (95% CI) of serious infection were 8.66 (2.81 to 20.22), 4.70 (2.63 to 7.75) and 8.07 (6.02 to 10.58) per 100 patient-years (/100pt-years) of follow-up in patients randomised to placebo, golimumab 50 mg and golimumab 100 mg, respectively. For patients treated with golimumab 50 mg, the incidence/100pt-years (95% CI) of serious infections was not increased at week 52 (5.78 (2.77 to 10.63)) or week 100 (5.79 (3.24 to 9.95)) but was increased for golimumab 100 mg at week 160 (8.07) relative to week 52 (6.10) and week 100 (6.63). However, the 95% CI at week 100 (4.33 to 9.72) and week 160 (6.02 to 10.58) was contained within that for week 52 (3.05 to 10.92) (table 4). In the golimumab 100 mg group, one patient had histoplasmosis and another had pulmonary tuberculosis. Further details of these opportunistic infections are provided in the online supplement.
The incidences (95% CI) of death through week 160 were 1.73 (0.04 to 9.65), 0.00 (0.00 to 0.94) and 0.62 (0.17 to 1.59)/100pt-years, respectively, for placebo, golimumab 50 mg and golimumab 100 mg (table 4). As reported previously,1 one placebo-treated patient died 6 months after enrolment. This 80-year-old woman who did not EE and never received golimumab died from pancreatic cancer on day 177. Four additional patients, all women who received golimumab 100 mg, died between weeks 100 and 160. The reported causes (and times) of death were aggressive lymphoma (week 132), cardiovascular event (week 141), congestive heart failure (week 146) and pneumonia (week 158). Further details of patient deaths are provided in the online supplement.
The incidences (95% CI) of malignancy at week 160 were 1.73 (0.04 to 9.66), 0.95 (0.20 to 2.77) and 2.04 (1.09 to 3.49)/100pt-years for placebo, golimumab 50 mg and golimumab 100 mg, respectively. The largest difference between groups was observed for lymphoma, with incidences (95% CI) of 0.00 (0.00 to 0.94) for golimumab 50 mg and 0.62 (0.17 to 1.59) for golimumab 100 mg (table 4). Among patients who received golimumab 100 mg, although the incidence (95% CI) of malignancy/100pt-years was not increased from week 52 (1.12 (0.14 to 4.03)) to week 100 (0.77 (0.16 to 2.24)), it was higher at week 160 (2.04 (1.09 to 3.49)).
In an analysis comparing malignancy incidences with expected rates in the general US population per the SEER database (table 5), the 95% CI of SIRs for all treatment groups included unity. Thus, the incidence of all malignancies occurring among patients treated in this study does not differ significantly from those expected in the general US population. However, the 95% CI of the SIR for lymphoma in the golimumab 100 mg group (5.55 to 52.15) did not include unity, suggesting that the lymphoma risk is increased with golimumab 100 mg (table 5).
Number of patients with one or more malignancies through week 160 compared with the expected number of malignancies from the general US population according to the SEER database
Discussion
We previously reported on the use of subcutaneous golimumab in 461 patients with active RA who have previous experience with TNF antagonists in GO-AFTER, the first prospective, randomised, double-blind, placebo-controlled trial conducted in this patient population and the only such study with efficacy analysed according to randomised treatment groups. GO-AFTER has the longest planned follow-up period (5 years) among studies involving similar patient populations. Through week 24, golimumab 50 mg and 100 mg every 4 weeks yielded statistically significantly higher proportions of patients achieving clinical response and clinically meaningful improvements in physical function versus placebo and no unexpected safety concerns.1
The GO-AFTER LTE that began at week 24 is ongoing; we now report findings through week 160, during which patients randomised to placebo crossed over to golimumab 50 mg and then all patients receiving golimumab 50 mg could have escalated from 50 mg to 100 mg. Patients randomised to golimumab 100 mg did not change treatment. Although comparative efficacy of golimumab 50 mg and 100 mg was similar at week 14, the golimumab dose was increased for 70% of eligible patients during the LTE, likely reflecting the refractory nature of disease in these patients who have previous experience with TNF inhibitors. Alternatively, the durability of clinical response to golimumab 50 mg may be less than that to golimumab 100 mg in this patient population beyond 24 weeks or it could be that investigator discretion, rather than objective escalation criteria, allowed for the golimumab dose to be escalated despite achievement of an otherwise acceptable response.
The clinical response observed through week 24 was reproduced in the LTE within the placebo group, whereby similar ACR20/50/70 response rates were observed after switching to golimumab. Moreover, improvements in clinical signs/symptoms and physical function observed through week 241 were maintained or enhanced through week 100 for most patients remaining in the trial. Importantly, 40–45% of patients who remained in the study sustained a DAS28-CRP score <3.2 through week 160. Golimumab dose escalation to 100 mg during the LTE increased clinical response rates from before to after dose escalation, suggesting that patients who have previous experience with TNF inhibitors can benefit from golimumab dose increase(s).
The ability to draw firm conclusions from these dose escalation data, however, is limited by several factors, including the following: (1) relatively small numbers of patients were available for analysis at later follow-up visits, (2) the trial was neither designed nor powered to compare treatment groups beyond week 24 and (3) dose escalation was at the sole discretion of investigators. Nonetheless, taken together, the demonstration of longer-term efficacy provides additional support for the week 24 observation that patients who previously discontinued TNF-antagonist treatment for any reason respond to golimumab.
Nearly 50% of randomised patients discontinued by week 160, which is higher than discontinuation rates observed in other phase 3 golimumab trials, that is, 23% and 26% in GO-BEFORE and GO-FORWARD by week 160 (data not shown). This observation, as well as that of unsatisfactory therapeutic effect and AEs being common discontinuation reasons, is not surprising given that a total of 35% of these patients who have previous experience with TNF inhibitors (>65% of whom also have previous experience with synthetic DMARD, with ‘synthetic DMARDs’) discontinued such prior treatment due to intolerance (63/445 patients) or unsatisfactory therapeutic effect (92/445 patients) and thus represent a population enriched with patients refractory to and/or intolerant of such treatment. Approximately two-thirds of patients continued pre-existing methotrexate treatment; the remaining patients received golimumab monotherapy. The length of the GO-AFTER LTE must also be considered when interpreting findings related to study agent discontinuation and efficacy (see above).
The short-term golimumab safety profile in patients who have previous experience with TNF inhibitors was similar to that for placebo throughout the 16-week placebo-controlled period.1 Through week 160, the length of follow-up for the placebo-controlled phase was 16 weeks versus 60 and 102 weeks for golimumab 50 mg and 100 mg, respectively. Thus, the higher incidences observed for specific AE categories through week 160 (table 4) relative to observations through week 161 were not unexpected given follow-up periods almost 4 times (50 mg) and more than 6 times (100 mg) the length of placebo-controlled follow-up. Also through week 160, patients treated with golimumab 100 mg received an average cumulative dose >3 times that for golimumab 50 mg (table 4), yielding an overall follow-up of golimumab 100 mg (645 pt-years) ∼10 times that for placebo (58 pt-years) and 2 times that for golimumab 50 mg (319 pt-yrs). These imbalances in exposure/follow-up were largely driven by the fact that, while 33% of enrolled patients were randomised to golimumab 100 mg, by week 160, 81% of the remaining patients were receiving golimumab 100 mg. When adjusted for length of patient follow-up, however, the incidences of serious infection, malignancy/lymphoma and death were higher in the 100 mg group, indicating a potential effect of golimumab dose. A similar pattern of a higher incidence of certain AEs with higher drug exposure was observed in the START trial of infliximab in patients with RA. In that trial, high serum infliximab concentrations during the induction regimen of 10 mg/kg given at weeks 0, 2 and 6 may have been associated with an increased risk of serious infections during the first 22 weeks of the trial. During the maintenance period, when infusions were administered every 8 weeks and peak serum infliximab concentrations may not have reached the same level as they did during the induction period, the risk of serious infection was reduced and consistent with that observed with lower infliximab doses.14
B cell lymphomas were the most common of lymphoma cases (supplementary table 1), which is consistent with that observed in patients with RA.15 Irrespective of TNF-antagonist treatment, patients with RA have an increased lymphoma risk versus the general population;16 this risk increases with disease duration and severity.15 ,17,–,19 GO-AFTER patients, with average disease durations of 10.6–12.4 years and previous receipt of anti-TNF treatment (indicating active refractory disease), represent an RA cohort consistent with increased lymphoma risk.15
The potential contribution of anti-TNF treatment to lymphoma remains an area of debate. Although an increased risk has been suggested with anti-TNF treatment,20 an alternative hypothesis asserts that lymphoma risk is actually related to RA severity and may decrease with control of chronic inflammation via disease-modifying treatment.18 ,21 Consistent with this hypothesis, Askling and colleagues22 observed no overall elevation of cancer risk and no increase with increasing duration of anti-TNF treatment in their evaluation of 6366 patients with RA (25 693 pt-years of follow-up). Data supporting the possible relationship between clinical status and lymphoma have also been reported by Wolfe and Michaud in their prospective evaluation of 19 562 patients (89 710 pt-years) enrolled in a longitudinal RA registry.23 ,24 However, because patients with the worst clinical status appeared to receive anti-TNF treatment preferentially, the authors were limited in their ability to establish causal relationships between RA treatment and lymphoma.23 ,24
At baseline, the four GO-AFTER patients diagnosed as having lymphoma had disease durations ranging from 5 to 31 years and substantial tender/swollen joint counts; three of the four patients also had baseline DAS28 and/or SDAI scores falling within the higher quartiles of scores observed across all patients, and follow-up scores at the time of lymphoma diagnosis were largely consistent with those documented at baseline, with the exception of one patient whose DAS28 and SDAI scores improved from a higher to a lower quartile from baseline to that time. Further discussion of these four patients with lymphoma is provided as online supplemental text.
In addition to the study limitations discussed above, the ability to evaluate effects of golimumab dose on safety was also limited by the GO-AFTER study design that allowed for dose escalation and yielded markedly differing lengths of follow-up for golimumab 50 mg (319 pt-years) versus 100 mg (645 pt-years).
Taken together with findings previously reported through week 24 of the GO-AFTER trial,1 these longer-term findings through 3 years support the efficacy of golimumab in patients with active RA previously treated with TNF antagonist(s); definitive conclusions regarding long-term safety will require further monitoring.
Acknowledgments
The authors thank Mahboob U Rahman, MD (a former Janssen employee who is now an employee of Pfizer Inc.), for his work pertaining to the GO-AFTER clinical trial and Michelle L Perate, MS, and Mary H Whitman, PhD, of Janssen Biotech Inc. for their excellent assistance with manuscript preparation and submission.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Web Only Data - This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
-
Funding This study was sponsored by Janssen Research & Development and Merck/Schering-Plough Research Institute.
-
Competing interests JS Smolen has received research grant support from Abbott, BMS, MSD, Pfizer, Roche and UCB and consultation and/or speaking honoraria from Abbott, Astra-Zeneca, BMS, Celgene, Glaxo, Janssen, MSD, Novo-Nordisk, Pfizer, Roche, Sanofi-Aventis and UCB. J Kay has received research funding paid to the University of Massachusetts Medical School from Bristol Myers Squibb Co., F. Hoffmann-La Roche Ltd. and Sanofi-Aventis and consulting income from Bristol Myers Squibb Co., Crescendo BioScience Inc., Eisai Co. Ltd., Janssen, Johnson & Johnson, Mallinckrodt Inc., NovoNordisk Inc., Pfizer Inc. and UCB S.A. R Landewé has received research grant support from Abbott, Pfizer, Roche and UCB and consultation and/or speaking honoraria from Abbott, Astra-Zeneca, BMS, Glaxo, Janssen, MSD, Pfizer, Roche and UCB. EL Matteson has received research grant support and consultation honoraria from Janssen. N Gaylis has received research grant support and consultation and/or speaking honoraria from Janssen/Johnson & Johnson and serves as Medical Director Rheumatology Division—Cardinal Health. J Wollenhaupt has received consultation and/or speaking honoraria from Abbott, Amgen, BMS, Chugai, MSD, Medac, Pfizer, Roche, Sanofi-Aventis and UCB. FT Murphy has received speaking honoraria from Abbott Immunology and Janssen. Y Zhou, EC Hsia and MK Doyle are employees of Janssen Research and Development.
-
Ethics approval All patients provided written informed consent, and the protocol was approved by each institution's human subjects ethical review board.
-
Provenance and peer review Not commissioned; externally peer reviewed.