Article Text

Abstract
Background Type I interferons (IFNs) appear to play a central role in disease pathogenesis in systemic lupus erythematosus (SLE), making them potential therapeutic targets.
Methods Safety profile, pharmacokinetics, immunogenicity, pharmacodynamics and clinical activity of sifalimumab, an anti-IFNα monoclonal antibody, were assessed in a phase I, multicentre, randomised, double-blind, dose-escalation study with an open-label extension in adults with moderately active SLE. Subjects received one intravenous dose of sifalimumab (n=33 blinded phase, 0.3, 1, 3, 10 or 30 mg/kg; n=17 open-label, 1, 3, 10 or 30 mg/kg) or placebo (n=17). Each phase lasted 84 days.
Results Adverse events (AEs) were similar between groups; about 97% of AEs were grade 1 or 2. All grade 3 and 4 AEs and all serious AEs (2 placebo, 1 sifalimumab) were deemed unrelated to the study drug. No increase in viral infections or reactivation was observed. Sifalimumab caused dose-dependent inhibition of type I IFN-induced mRNAs (type I IFN signature) in whole blood and corresponding changes in related proteins in affected skin. Exploratory analyses showed consistent trends toward improvement in disease activity in sifalimumab-treated versus placebo-treated subjects. A lower proportion of sifalimumab-treated subjects required new or increased immunosuppressive treatments (12% vs 41%; p=0.03) and had fewer Systemic Lupus Erythematosus Disease Activity Index flares (3% vs 29%; p=0.014).
Conclusions Sifalimumab had a safety profile that supports further clinical development. This trial demonstrated that overexpression of type I IFN signature in SLE is at least partly driven by IFNα, and exploratory analyses suggest that IFNα inhibition may be associated with clinical benefit in SLE.
Trial registration number NCT00299819.
Statistics from Altmetric.com
Introduction
Systemic lupus erythematosus (SLE) is a chronic, heterogeneous, autoimmune disease which can affect almost any organ in the body.1 2 Active disease impairs quality of life and leads to irreversible organ damage, long-term morbidity and mortality.3,–,8 Hospitalisations9 and side effects of chronic corticosteroids and other immunosuppressive treatments add to the disease burden.10 11 There is a significant unmet need for the development of safer and more effective treatments, providing a rationale for the study of novel biological agents targeting pathways important in the perpetuation of this disease.
Type I interferons (IFNs), including IFNα, are implicated in SLE pathogenesis.12 13 Heritability of high serum IFNα activity,14 and genetic polymorphisms affecting type I IFN production, activity, signalling, or induced proteins have been associated with susceptibility to SLE.15,–,19 Immune complexes containing anti-double-stranded DNA (anti-dsDNA) antibodies and nucleic acids act as potent inducers of IFNα.20,–,26 Elevated IFNα-producing cells, IFNα mRNA and IFNα protein, as well as mRNAs and proteins induced by type I IFNs (type I IFN signature) have been reported in blood or tissue of patients with SLE.27,–,39 Exogenous type I IFNs can also induce or worsen clinical manifestations of SLE.40,–,42
Sifalimumab (formerly MEDI-545; MedImmune, LLC, Gaithersburg, Maryland, USA) is a fully human IgG1κ monoclonal antibody that binds to IFNα with high affinity and prevents IFNα signalling through its receptor, IFNAR.
In non-clinical experiments, sifalimumab has been shown to neutralise a spectrum of IFNα subtypes in Daudi cell proliferation assays; decrease IFNα-induced cell surface and soluble markers in healthy donors; and exhibit high affinity for antigen (unpublished data).
In this study, the safety profile, pharmacokinetics and immunogenicity of a single intravenous dose of sifalimumab were examined and exploratory analyses of the effects of sifalimumab on pharmacodynamic markers and disease activity carried out.
Subjects and methods
Study design
Study MI-CP126 was a phase I, randomised, double-blind, placebo-controlled, single-dose, dose-escalation, multicentre study with an open-label phase designed to examine the safety profile and tolerability of sifalimumab (primary objective) and its pharmacokinetics and immunogenicity (secondary objectives). Exploratory analyses included evaluating the effects of sifalimumab on pharmacodynamic markers and SLE activity. This study was conducted according to the Declaration of Helsinki, with protocol approval by the institutional review boards at participating sites. Subjects gave written informed consent before the study.
Adults aged ≥18 years who met four or more of the 11 revised American College of Rheumatology classification criteria for SLE43 44 were randomised 2:1 to receive one intravenous dose of sifalimumab (0.3, 1, 3, 10 or 30 mg/kg) or placebo, in ascending dose, blinded cohorts. In each cohort, six to eight subjects received sifalimumab and three to four received placebo on study day 0. After all subjects had been randomised into the blinded phase of the study, additional subjects were enrolled and received sifalimumab in an open-label extension. These included new subjects and subjects from the blinded phase who had previously received placebo. The new subjects were sequentially entered to receive sifalimumab at single intravenous doses of 1, 3 or 10 mg/kg. The placebo subjects from the blinded phase had the option of entering the open-label extension only after unblinding of their treatment assignment; those who entered the open-label phase received sifalimumab at 30 mg/kg. In each phase, subjects were monitored for 84 days after dosing.
Paracetamol, non-steroidal anti-inflammatory drugs, antimalarial agents and prednisone (≤20 mg/day or equivalent) were allowed if the regimen had remained unchanged for ≥28 days before randomisation (study day 0). Subjects were excluded if they had received cyclophosphamide, other immunosuppressant agents, immunoglobulins, blood products, antiviral treatment, or an investigational treatment within 28 days of study day 0. Other exclusions included history of primary immunodeficiency, cancer, stroke, severe herpes infection; active or chronic viral infections; active central nervous system lupus; and clinically significant cardiac, liver or renal disease.
Safety profile
Adverse events (AEs) and safety laboratory assessments were monitored on days 0, 1, 2, 4, 7, 14, 21, 28, 42, 56 and 84. AEs and serious AEs (SAEs) were graded on a scale of 1–5 using the National Cancer Institute Common Terminology Criteria for Adverse Events, version 3.045 and by relationship to the study drug. AEs were summarised by organ class and preferred terms (Medical Dictionary for Regulatory Activities, version 11).46 AEs were defined as unfavourable events occurring after study drug administration, whether or not they were considered treatment related. SAEs were AEs that resulted in death, were life-threatening, required or prolonged existing hospitalisation and/or resulted in significant disability or congenital abnormality. SLE flare, if present as determined by the doctor, was reported as an AE (ie, increased SLE disease activity). A flare could be detected at scheduled visits or at any other time during the study.
Serum chemistry, complete blood count with differential and urine analysis were assessed throughout the study. New abnormal laboratory test results were reported as AEs regardless of clinical significance.
Viral monitoring was performed on days 0 (before dosing), 7, 14, 28 and 84. Surveillance was performed for Epstein–Barr virus (EBV), cytomegalovirus (CMV), herpes simplex virus (HSV-1 and HSV-2) and human papilloma virus (HPV). Oropharyngeal swabs were cultured for HSV-1 and -2, and vaginal swabs from women were cultured for HSV-2 and tested for HPV DNA. Copy numbers of EBV and CMV DNAs were assessed in blood samples by PCR.
Immunogenicity
Anti-sifalimumab antibodies were tested on study days 0 (before dosing), 14, 28, 42 and 84 (see online supplementary text for assay details).
Pharmacokinetics
Sifalimumab concentrations and pharmacokinetic parameters were evaluated before and after dosing (see online supplementary text file for details).
Pharmacodynamics
The expression level of type I IFN-inducible mRNAs (type I IFN signature) in whole blood was measured by TaqMan Low Density Array (quantitative reverse transcriptase-PCR (qRT-PCR)) (Applied Biosystems, Foster City, California, USA).47 48 Blood and skin biopsy collection (before and after dosing) and measurement of type I IFN signature inhibition in whole blood were performed as published27 (see online supplementary text file for details).
Immunohistochemistry was used to evaluate expression of two IFNα/β-inducible proteins: ubiquitin-specific peptidase 18 (USP18) and epithelial stromal interaction 1 (ESI-1), selected because corresponding mRNA levels were elevated in affected skin (see online supplementary text file for methods).
Clinical activity
Disease activity was examined using the Safety of Estrogens in Lupus Erythematosus: National Assessment–Systemic Lupus Erythematosus Disease Activity Index (SELENA-SLEDAI) score, including a Physician's Global Assessment (PGA) 49; the British Isles Lupus Activity Group (BILAG) index50 51; initiation or increased immunosuppressant agents; serum C3 and anti-dsDNA antibody levels; and Krupp Fatigue Severity score.52 SLEDAI and BILAG were scored on study days 0, 14, 28 and 56 and study days 0, 28 and 56, respectively. Photographs of skin lesions were taken on study days 0, 7, 14, 28, 56 and 84. Skin lesions were graded on a scale of 0–4 (0, worsened; 1, unchanged; 2, improved; 3, nearly clear; 4, clear compared with study day 0) using the median score of three independent reviewers blinded to treatment assignment.
Statistics
No formal statistical calculation was performed to determine the necessary sample size for the blinded or open-label phase. For binary variables, groups were compared using the Fisher exact test; for continuous variables, analysis of variance was used unless noted otherwise. Change from baseline in Krupp Fatigue Severity score was compared between placebo and sifalimumab groups using analysis of covariance with adjustment to baseline. Reported p values are two-sided. p Values were not calculated for safety, pharmacokinetic, or immunogenicity data.
Results
Subject demographics and disposition
Sixty-nine subjects were enrolled in the blinded and open-label phases. The blinded phase included 51 subjects (sifalimumab, n=34; placebo, n=17) from 13 US sites. All subjects, except one (0.3 mg/kg sifalimumab group) who withdrew consent before dosing, received the study drug and completed follow-up (0.3 mg/kg, n=6; 1 mg/kg, n=6; 3 mg/kg, n=6; 10 mg/kg, n=7; 30 mg/kg, n=8). Eighteen subjects were enrolled in the open-label phase at six sites (three had previously received placebo in the blinded phase); 17 completed this phase (1 mg/kg, n=6; 3 mg/kg, n=4; 10 mg/kg, n=4; 30 mg/kg, n=3). One subject in the 10 mg/kg sifalimumab group withdrew consent before dosing.
Most subjects were Caucasian (78%), female (96%) and receiving antimalarial drugs (72%) for mildly to moderately active SLE (table 1). At baseline, one subject (placebo) had a SLEDAI score >10. Five subjects (three placebo, two sifalimumab) had a BILAG A score ≥1, indicating severe activity in one or more organs.
Demographics and selected baseline characteristics
Safety profile
During the blinded phase, 202 AEs were reported in 33 sifalimumab subjects and 126 in 17 placebo subjects. In the open-label phase, there were 102 AEs in 16 subjects. AE rates per subject were similar among groups during the blinded (placebo, 7.4; sifalimumab, 6.1) and open-label (6.0) phases. SLE flare was the most common AE in both groups during the blinded phase, occurring in a higher proportion of placebo subjects. Headache was the next most common AE in the blinded phase, occurring in a similar proportion of placebo and sifalimumab subjects (table 2).
Adverse events (AEs)*
There were 29 treatment-related AEs during the blinded phase: eight events in six placebo subjects and 21 events in eight sifalimumab subjects. Treatment-related AEs occurring in more than one subject were headache (n=4; one subject each in the placebo, 1 mg/kg, 10 mg/kg and 30 mg/kg groups), nausea (n=2, one subject each in the 1 and 30 mg/kg groups), increased alanine aminotransferase (n=2, both in the placebo group), pruritus (n=2, both in the 30 mg/kg group) and pruritic rash (n=2, one subject each in the placebo and 1 mg/kg groups).
Most AEs were mild; no subject withdrew owing to an AE. In the blinded phase, grade 1 and 2 AEs occurred in 72% and 25% of placebo subjects, respectively, and 85% and 13% of sifalimumab subjects. Six grade 3 AEs (placebo, n=4 (diverticulitis, post-traumatic pain, SLE flare, sciatica); sifalimumab 1 mg/kg, n=2 (decreased haemoglobin associated with menorrhagia, anaemia)) and one grade 4 AE (sifalimumab 30 mg/kg (transient elevation in blood potassium)) were reported. No grade 3 or 4 AEs were considered treatment related. In the open-label phase, two grade 3 AEs (one subject experienced two episodes of lymphopenia; each resolved without treatment) and no grade 4 AEs were reported. No relationship was apparent between sifalimumab dose and severity or frequency of AEs.
Three SAEs were reported (diverticulitis and pain related to traumatic hip dislocation in the placebo group; anaemia in the sifalimumab 1 mg/kg group). All were in the blinded phase and considered unrelated to treatment.
Infections occurred in 41% of placebo and 39% of sifalimumab subjects during the blinded phase; only nasopharyngitis (placebo, n=2; sifalimumab, n=3), upper respiratory tract infection (placebo, n=2; sifalimumab, n=3) and urinary tract infection (sifalimumab, n=3) occurred in more than one subject. Three infections were considered treatment related, one in placebo (oral herpes) and two in sifalimumab (sinusitis, upper respiratory tract infection) subjects. During the open-label phase, 35% of subjects had an infection. All infections were grade 1 or 2 in severity, except for one grade 3 diverticulitis in a placebo subject.
All viral reactivations were grade 1 in severity. During the blinded phase, one subject (6%) in the placebo group who had been receiving oral corticosteroid since day 15 had oral herpes on day 28; two subjects (6%) in the sifalimumab-treated groups had viral reactivations: one had oral herpes on study day 19 and one had herpes zoster on study day 84. The subject with herpes zoster received intramuscular corticosteroid (60 mg) on study day 56. During the open-label phase, one subject (6%) developed grade 2 genital herpes on study day 29.
Viral surveillance for CMV, EBV, HPV, HSV-1 and HSV-2 showed a low percentage of subjects converting from negative to positive test results during follow-up, and similar conversion rates in placebo and sifalimumab groups (table 3). The most common conversions were HPV high risk for cervical cancer, in two of nine placebo subjects (22%) and 2 of 16 sifalimumab subjects (13%). No time or dose relationships were observed for these events. Of those who had a conversion and at least one subsequent test, all but one converted back to negative. Of 13 subjects (placebo n=1; sifalimumab n=12) with viral evidence at baseline, the test results were negative in 10 subjects on subsequent testing after study drug administration. One subject with a positive test at baseline converted to negative at day 14 and converted back to positive on day 84; two subjects stayed positive through day 84. Nine (positive HPV high-risk test result, n=4; positive HPV low-risk test result, n=3; positive HSV-1 oropharyngeal test result, n=2) of the 10 positive-to-negative conversions were in subjects who received sifalimumab.
Follow-up viral surveillance testing
No study-related deaths, drug administration interruptions, anaphylactic or anaphylactoid reactions, cases of cytokine release syndrome, or new immune complex diseases were reported. AEs that might indicate allergic reactions to study drug included one hypersensitivity AE (sifalimumab) and seven skin AEs (including pruritus, generalised pruritus, papular rash, pruritic rash), which occurred in three (17.6%) placebo and four (12.1%) sifalimumab subjects.
Immunogenicity
All subjects in both phases tested negative for anti-sifalimumab antibodies at baseline and none developed a detectable binding antibody to sifalimumab.
Pharmacokinetics
During the blinded phase, mean serum sifalimumab concentrations after a single intravenous dose exhibited a shallow initial distribution phase followed by a prolonged terminal elimination phase (figure 1). The mean concentration–time profiles were similar for all dose groups with the exception of the 1 mg/kg group, for which outlier concentrations in one subject resulted in a longer half life compared with other doses. Mean serum sifalimumab Cmax increased dose proportionally from 11 µg/ml after the 0.3 mg/kg dose to 1206 µg/ml after the 30 mg/kg dose. Individual variability in clearance ranged from 30% to 40% across the dose groups (except 1 mg/kg). Similar dose proportionality was observed for mean serum area under the curve (AUC0–∞) over the dose range. Mean serum sifalimumab T1/2 was 15–24 days across different dose levels except for the 1 mg/kg dose group, which was 34 days. Mean total body serum CL (clearance) ranged between 143 and 197 ml/day across the dose groups. Mean values for the Vdss and Vdz were similar and low, about 3−7 litres suggesting limited distribution of sifalimumab systemically.

Mean serum concentrations after a single intravenous dose of sifalimumab (0.3 (n=6); 1 (n=6); 3 (n=6); 10 (n=7); 30 (n=8) mg/kg) in subjects with SLE (blinded phase of the study). Blood was collected at multiple times up to day 84. Serum concentrations of sifalimumab were measured using a qualified ELISA; the assay was linear in the range of 1.25 μg/ml to 40.0 μg/ml. Individual variability in clearance ranged from 30% to 40% across the dose groups (except 1 mg/kg). SLE, systemic lupus erythematosus.
Pharmacodynamics
As previously reported using microarrays,27 and confirmed here with TaqMan data, subjects with an elevated baseline type I IFN signature (n=36 (58%) of 62 tested) who received sifalimumab had dose-dependent inhibition of the signature (figure 2). The 0.3, 10 and 30 mg/kg groups were statistically different from placebo (p=0.05, 0.01 and 0.01; Hotelling's T2 test with pairwise comparisons up to 14 days after dosing, which is the approximate T1/2 of sifalimumab). Type I IFN signature in subjects treated with 1 or 3 mg/kg sifalimumab tended to be different from those in placebo subjects after treatment (p=0.09 and 0.06, respectively).

Effects of sifalimumab on type I IFN gene signature in subjects with SLE. Level of expression of 21 IFNα/β inducible genes for each subject relative to baseline expression. For this plot, SLE subjects must have had an overexpression of the gene signature of at least threefold that were normalised to 1 at baseline. The change from baseline mean±SE level of signature gene expression in whole blood after sifalimumab treatment is shown. Maximum neutralisation of the gene signature occurred within 24 h after treatment. Hotelling's T2 test with pairwise comparisons up to 14 days after dosing was used for this analysis. IFN, interferon; SLE, systemic lupus erythematosus.
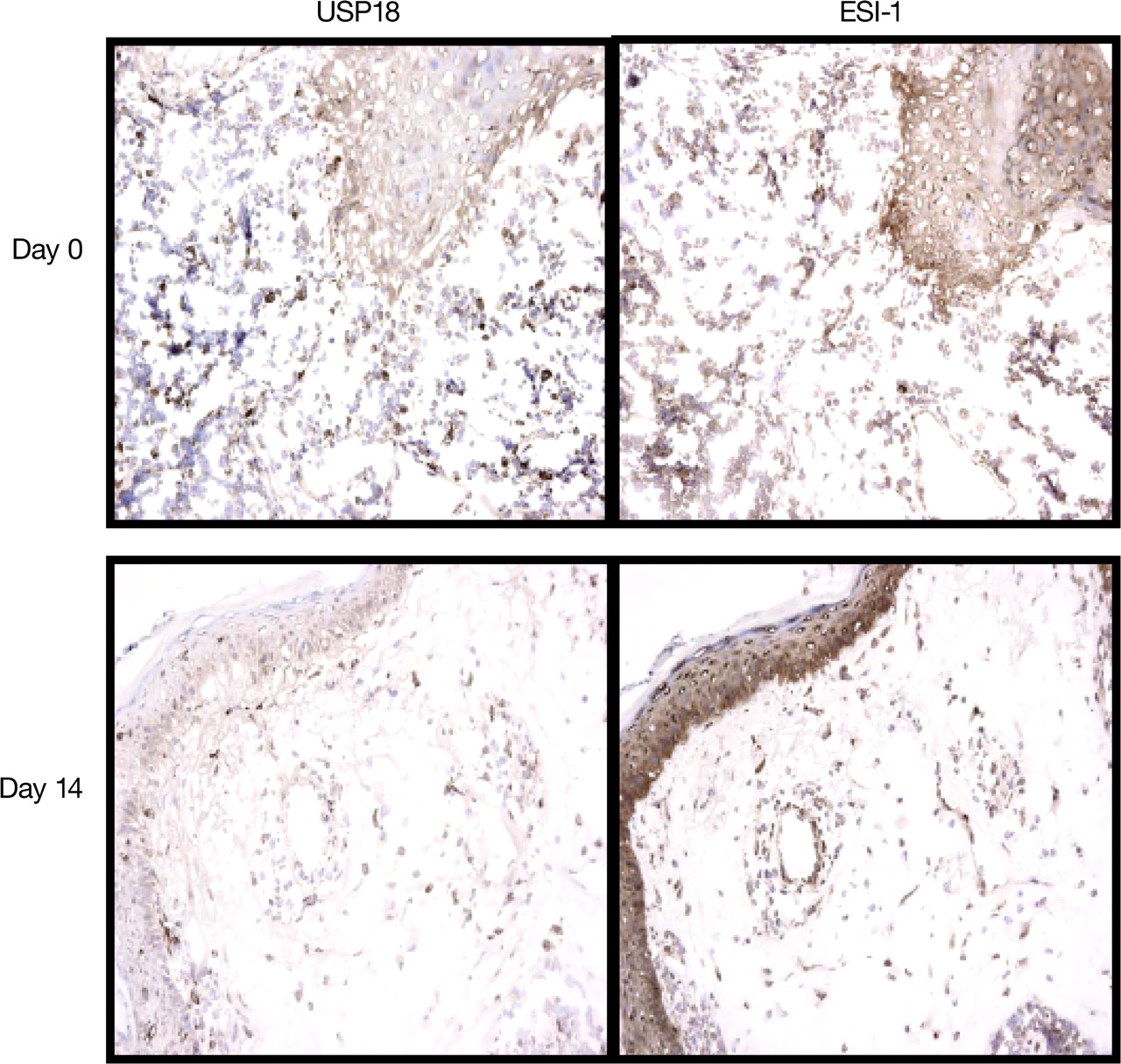
The effect of sifalimumab (10 mg/kg) on IFNα/β-inducible proteins in paired skin biopsy samples from a subject with SLE is depicted in figure 3. USP18 and ESI-1 protein overexpression, observed in lesional skin at study day 0, was decreased on study day 14 after receiving the dose.

Effect of sifalimumab treatment on IFNα/β inducible proteins in skin biopsy samples from a subject with SLE. Immunohistochemical analyses of paired biopsy specimens (day 0 before dosing and day 14 after dosing) from lesional skin obtained from a subject treated with 10 mg/kg of sifalimumab. The ESI-1 and USP18 protein overexpression that was observed (as indicated by dark staining) in lesional skin at study day 0 was decreased in the skin at study day 14 after dosing. ESI-1, epithelial stromal interaction 1; IFN, interferon; SLE, systemic lupus erythematosus; USP18, ubiquitin-specific peptidase 18.
Clinical activity
In the blinded phase, an exploratory analysis of the effects on disease activity of sifalimumab (all doses combined) compared with placebo was carried out. Consistent trends of greater improvement in the sifalimumab group were found (figure 4) using different measures, although statistical significance was not usually attained. More sifalimumab subjects achieved mild disease, defined by a PGA score of ≤0.5 on a three-inch Visual Analogue Scale at any time after day 0 (figure 4A), and achieved inactive disease, with a SLEDAI score of 0 at any time after day 0 (figure 4B). The adjusted mean SLEDAI score, normalised to 1 on study day 0 for each subject, was reduced over time in sifalimumab subjects but increased over time in placebo subjects (figure 4C). Among subjects who had a SLEDAI score ≥4 on study day 0, more sifalimumab subjects improved in SLEDAI by four or more points (figure 4D) at any time after day 0 and achieved a composite response of reduction in SLEDAI score ≥4 points, no new BILAG score of B or more, and no PGA worsening of >0.3 from baseline at any time after day 0 (figure 4E).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Clinical effects of sifalimumab on improvement in disease activity and lupus flares (blinded phase). Fisher's exact test was used for all statistical analyses unless indicated otherwise. (A) Percentage of subjects who achieved mild disease, as assessed by a PGA score of ≤0.5 on a three-inch Visual Analogue Scale (p=0.108 at any time after day 0; n=16 placebo, n=30 sifalimumab). (B) Percentage of subjects with baseline SLEDAI>0 who achieved inactive disease, as assessed by a SLEDAI score of 0 (p=0.095 at any time after day 0; n=17 placebo, n=31 sifalimumab). (C) Adjusted mean SLEDAI score over time (p=0.105 for study day 14, p=0.070 for study day 28 and *p=0.030 for study day 56 based on analysis of variance; n=17 placebo, n=31 sifalimumab). The adjusted mean SLEDAI was developed by computing the area under the curve of the SLEDAI global score over time divided by time interval. The adjusted mean SLEDAI was further normalised to 1 at baseline and summarised by treatment group at study days 14, 28 and 56. (D) Percentage of subjects with baseline SLEDAI score ≥4 who achieved an improvement in SLEDAI of ≥4 points (p=0.143 at any time after day 0; n=13 placebo, n=23 sifalimumab). (E) Percentage of subjects who achieved a composite response of reduction in SLEDAI score ≥4 points, no new BILAG A score and no new BILAG B score, and no PGA worsening of >0.3 from baseline (p=0.438 at any time after day 0; n=13 placebo, n=23 sifalimumab). (F) Percentage of subjects who did not have a SLEDAI flare at study day 0 and who subsequently experienced a SLEDAI flare, defined as a ≥4-point increase from baseline (study day 0) in SLEDAI score (*p=0.014 at any time after day 0; n=5 placebo, n=1 sifalimumab), (G) Percentage of subjects who experienced a BILAG flare, as assessed by worsening of the BILAG score with at least one new BILAG B score (p=0.294 at any time after day 0; n=17 placebo, n=33 sifalimumab). (H) Percentage of subjects who required new or increased lupus treatment at any time during the study (*p=0.03; n=17 placebo, n=33 sifalimumab). BILAG, British Isles Lupus Activity Group; PGA, Physician's Global Assessment; SLEDAI, Systemic Lupus Erythematosus Disease Activity Index.
A smaller proportion of sifalimumab subjects had worsening of disease compared with placebo. Sifalimumab subjects were less likely to exhibit a SLEDAI flare (SLEDAI increase four or more points at any time after day 0) (figure 4F),53 or a BILAG flare (one or more new BILAG B) (figure 4G). New or increased treatment for SLE, including antimalarial agents and/or corticosteroids was less in sifalimumab subjects than placebo subjects (figure 4H). Similar but less pronounced clinical effects of sifalimumab were observed in the open-label phase.
No effect of sifalimumab on the Krupp Fatigue Severity score was observed in either phase. Too few subjects had abnormal C3 and anti-DNA antibody levels at baseline (n=5 each) to compare these end points. Photography of target lesions was available in eight subjects in the blinded phase, seven of whom were in the sifalimumab group. Five of seven sifalimumab subjects had improving lesions on study days 14, 56, and 84, and four of six had improving lesions on study day 28. The target lesion in the placebo subject was clear on study day 14, but worse on study days 28, 56 and 84.
Discussion
SLE is now being targeted with a myriad of promising investigational treatments. Biological approaches to treating SLE currently include monoclonal antibodies, fusion proteins and peptides that block molecules or inhibit/kill cells thought to be involved in SLE pathogenesis.
In this first study of sifalimumab in human subjects with SLE, the primary objective was to assess its safety profile, including general effects, infusion or allergic reactions and potential risks related to inhibition of IFNα such as viral infections or viral reactivation. Most AEs were mild or moderate and expected in an SLE population. Although no serious allergic or infusion reactions to the study drug occurred, in another phase I study of sifalimumab in subjects with psoriasis, one serious infusion reaction was seen, thought to be related to rapid infusion.54
A potentially increased susceptibility to infections, including acute viral illness or reactivated latent infections, may accompany blockade of type I IFN signalling. In this study, placebo and sifalimumab subjects had similar rates of infections and diseases associated with viral reactivation. All infections in sifalimumab-treated subjects were grade 1 or grade 2 in severity. Proportions of subjects who had negative viral surveillance test results at baseline and then changed to positive results were similar in placebo-treated and sifalimumab-treated subjects. When follow-up testing was available, most subjects whose test results changed to positive subsequently reverted to negative, and some positive viral test results at baseline also reverted to negative after sifalimumab treatment. Together, these data do not suggest an increased risk of viral infection; however, this potential risk might become more apparent when larger numbers of subjects are exposed to sifalimumab for longer periods of time.
The single-dose pharmacokinetics of sifalimumab was linear and dose proportional across the dose range studied, with a mean half-life of about 15–24 days. Sifalimumab pharmacokinetic parameter values were typical of a monoclonal antibody without a significant target sink. Anti-sifalimumab antibodies were not detected, but the potential immunogenicity of sifalimumab requires further assessment in multiple-dose studies, using immunoassays capable of detecting antidrug antibody in the presence of drug.
Using TaqMan qRT-PCR technology, we confirmed that sifalimumab neutralises overexpression of the type I IFN signature in subjects with SLE in a dose-dependent manner. These data agreed with those generated by microarray technology, validating results previously published by Yao et al.27 Given the differences in sensitivities between the two assays, it was important to demonstrate that the IFN gene expression patterns were consistent using either technology.
Because there is overlap in the mRNAs induced by IFNα and IFNβ, the fact that sifalimumab significantly inhibited the type I IFN signature provides evidence that IFNα is a key contributor to this signature. These results, however, do not rule out the possibility of a role for IFNβ or other type I IFNs in the activation of type I IFN pathways in SLE.
Dermal inflammation and certain proteins associated with IFNα/β inducible gene expression (HERC5, ISG15, IP10) were previously found to decrease in the skin of sifalimumab-treated subjects within 2 weeks of dosing.27 In this study, we showed that USP18 and ESI-1 were also inhibited by sifalimumab, providing further evidence of a correlation between decreased IFN-inducible protein/gene expression and skin lesion improvement after a dose of sifalimumab.
In exploratory analyses, a single dose of sifalimumab was associated with trends toward improvement in disease activity and reduction in disease worsening across several measures. The adjusted mean SLEDAI scores demonstrated a statistically significant improvement in disease activity in sifalimumab subjects at study day 56.
In conclusion, a single intravenous administration of sifalimumab was not associated with any SAEs, produced a dose-dependent neutralisation of type I IFN gene signature, and suggested potential improvements in SLE disease. Our study supports the hypothesis that type I IFN may play an important role in SLE pathogenesis and that blockade of IFNα may be a useful strategy for reducing disease activity. Data on the single-dose testing of small numbers of subjects in a phase I trial have limitations. Repeated and longer exposure in subjects with more active disease, and studies powered to show efficacy, are needed to better understand the safety and potential clinical usefulness of inhibition of IFNα with sifalimumab.
Acknowledgments
In addition to the authors, the LISA study investigators include Stephen Bookbinder, Ocala Rheumatology Research Center, Ocala, Florida, USA; Carin Dugowson, University of Washington, Seattle, Washington, USA; Raymond Hausch, St Mary's Duluth Clinic, Duluth, Minnesota, USA; Kenneth Kalunian, University of California San Diego School of Medicine, La Jolla, California, USA; Robert Matheson, Oregon Medical Research Center, Portland, Oregon, USA; Frederick Murphy, Altoona Center for Clinical Research, Duncansville, Pennsylvania, USA; Yvonne Sherrer, Center for Rheumatology, Immunology, and Arthritis, Fort Lauderdale, Florida, USA. The authors thank Chris Morehouse and Brandon Higgs, MedImmune, LLC, for their technical input and Miriam Gitler, PhD, MedImmune, LLC, for her medical writing and editorial assistance in the preparation of the manuscript.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
Footnotes
JTM and DJW contributed equally to this study.
-
Funding This study and manuscript preparation were funded by MedImmune, LLC.
-
Competing interests JTM, DJW, MP, KAK, RL, SMB, VC and NO received research funding from MedImmune. JTM has received consulting fees from MedImmune, Biogen Idec, Genentech and Roche. Louisiana State University Health Sciences Center has received research funding on behalf of SMB from Biogen Idec, Genentech and Roche. YY, WIW, GR, LR, CL and BJ are employees of MedImmune. BW is a former employee of MedImmune.
-
Ethics approval This study was conducted with the approval of the institutional review boards at participating sites.
-
Provenance and peer review Not commissioned; externally peer reviewed.