Article Text

Abstract
Objectives: To evaluate efficacy, dose response, safety, and tolerability of adalimumab (D2E7) in disease modifying antirheumatic drug (DMARD) refractory patients with longstanding, active rheumatoid arthritis (RA).
Methods: During a 12 week, double blind, placebo controlled study, 284 patients were randomly allocated to receive weekly subcutaneous injections of adalimumab 20 mg (n = 72), 40 mg (n = 70), or 80 mg (n = 72) or placebo (n = 70) without concomitant DMARDs.
Results: Adalimumab significantly improved the signs and symptoms of RA for all efficacy measures. ACR20 responses with adalimumab were significant at each assessment versus placebo (p⩽0.01). Additionally, ACR responses with adalimumab were achieved more rapidly than with placebo, with 82/115 (71%) of the ultimate ACR20 response rate to adalimumab treatment achieved at week 2. At week 12, for adalimumab 20, 40, and 80 mg, ACR20 response rates were 50.7%, 57.1%, and 54.2%, respectively, versus 10.0% for placebo (p⩽0.001 for all comparisons); ACR50 rates were 23.9%, 27.1%, and 19.4%, respectively, versus 1.4% for placebo (p⩽0.001 for all comparisons); and ACR70 rates were 11.3%, 10.0%, and 8.3%, respectively, versus 0.0% for placebo (p⩽0.05 for all comparisons). All adalimumab doses significantly improved all ACR core criteria at all assessments. The 40 mg and 80 mg doses provided similar benefit. Adalimumab at all doses was generally well tolerated, with only mild or moderate adverse events. Completion rates were 87% for adalimumab and 67% for placebo.
Conclusions: Adalimumab given as monotreatment to patients with longstanding, severe RA refractory to traditional DMARDs produced a rapid, sustained response and was safe and well tolerated, with no dose limiting side effects.
- adalimumab
- monoclonal antibodies
- tumour necrosis factor α
- rheumatoid arthritis
- disease modifying antirheumatic drugs
- ACR, American College of Rheumatology
- AEs, adverse events
- ANAs, antinuclear antibodies
- ANCOVA, analysis of covariance
- CRP, C reactive protein
- CTC, Common Toxicity Criteria
- DAS, disease activity score
- DMARDs, disease modifying antirheumatic drugs
- dsDNA, double stranded DNA
- ESR, erythrocyte sedimentation rate
- HAQ, Health Assessment Questionnaire
- ITT, intention to treat
- MTX, methotrexate
- NSAIDs, non-steroidal anti-inflammatory drugs
- RA, rheumatoid arthritis
- sc, subcutaneously
- SJC, swollen joint count
- TJC, tender joint count
- TNFα, tumour necrosis factor α
Statistics from Altmetric.com
- adalimumab
- monoclonal antibodies
- tumour necrosis factor α
- rheumatoid arthritis
- disease modifying antirheumatic drugs
- ACR, American College of Rheumatology
- AEs, adverse events
- ANAs, antinuclear antibodies
- ANCOVA, analysis of covariance
- CRP, C reactive protein
- CTC, Common Toxicity Criteria
- DAS, disease activity score
- DMARDs, disease modifying antirheumatic drugs
- dsDNA, double stranded DNA
- ESR, erythrocyte sedimentation rate
- HAQ, Health Assessment Questionnaire
- ITT, intention to treat
- MTX, methotrexate
- NSAIDs, non-steroidal anti-inflammatory drugs
- RA, rheumatoid arthritis
- sc, subcutaneously
- SJC, swollen joint count
- TJC, tender joint count
- TNFα, tumour necrosis factor α
Rheumatoid arthritis (RA) is a chronic, systemic disorder that affects about 0.5–1.0% of adults in Western nations and is 2.5 times more prevalent among women than men.1,2 Characterised by progressive inflammatory synovitis, RA erodes articular cartilage and marginal bone and destroys joint structure and function,3 leaving patients with joint deformities, movement problems, and activity limitations. Within 10 years of disease onset, severe disability occurs in more than 50% of patients, and only 40–50% are able to work.4,5 After 20 years of persistent active RA, nearly 80% of people are partially disabled and 19% are completely disabled.6 In most cases, irreversible cartilage loss begins within a year of onset, with 70% of patients exhibiting radiographic disease progression after three years.7–9 For this reason, early diagnosis and aggressive treatment are now recognised as essential to the successful long term outcome of the disease.10–12
Although traditional treatments for RA such as non-steroidal anti-inflammatory drugs (NSAIDs) and corticosteroids have some benefit for temporary symptomatic relief,11 dramatic improvements in disease control have been noted with the use of traditional disease modifying antirheumatic drugs (DMARDs) such as methotrexate (MTX),13 which has been reported to slow the progression of joint destruction.14,15 Inadequate responders to MTX are often given MTX in combination with other agents, particularly hydroxychloroquine, sulfasalazine, leflunomide, and cyclosporin.15,16
In recent times, biological DMARD treatments for RA have been developed to inhibit the activity of proinflammatory cytokines such as tumour necrosis factor α (TNFα), which mediates the destructive synovitis that is the hallmark of RA. These agents have been successful in treating patients with RA who fail to respond adequately to traditional DMARD treatments or who cannot tolerate their toxic side effects.17–20 Two biological DMARDs designed to inhibit TNFα bioactivity have been approved by the European Medicine Evaluation Agency (EMEA) and the US Food and Drug Administration (FDA) for the treatment of RA: etanercept, a soluble TNF receptor—immunoglobulin G1 (IgG1) fusion protein (which also binds to TNFβ (lymphotoxin)),21–23 and infliximab, a chimeric (25% mouse/75% human) monoclonal antibody.24–27 Both agents are indicated for the treatment of moderate to severe DMARD refractory RA.28,29 Etanercept is administered subcutaneously (sc), whereas infliximab is given intravenously and only in combination with MTX.28,29
Adalimumab (D2E7; Abbott Laboratories, Abbott Park, Illinois, USA) is the first fully human anti-TNFα monoclonal antibody to enter clinical trials for the treatment of RA. Adalimumab is an IgG1, engineered using phage display technology with amino acid sequences from the human germ line, making it indistinguishable in structure and function from natural human IgG1.30–33 It has high specificity and affinity for TNFα (Kd = 6×10−10 M) but not TNFβ; it has a terminal half life comparable with that of human IgG1 (about two weeks).33,34 Additionally, adalimumab was developed to have a low degree of immunogenicity, enabling long term administration, with or without the concurrent use of immunosuppressant drugs such as MTX, and a low incidence of allergic reactions.33
Early phase I trials have demonstrated that adalimumab produces rapid and significant responses in patients with DMARD refractory active RA and suppresses long term radiological progression.35,36 Previously presented unpublished reports and published sources suggest that adalimumab may be effective both as monotreatment and as combination treatment with MTX for the treatment of RA.17–18,37,38,39,40,41,42,43,44,45,46 The current trial was conducted to evaluate the efficacy, dose response, safety, and tolerability of sc injections of adalimumab given weekly as monotreatment in patients with longstanding, active RA who were refractory to previous traditional DMARD treatment.
METHODS
Protocol
This phase II study was a 12 week, double blind, placebo controlled, multicentre trial conducted at 25 sites across nine European countries in accordance with the Good Clinical Practice Guidelines and principles of the Declaration of Helsinki. The ethics committee at each study site approved the study protocol. Patients were required to provide written informed consent.
After being screened for eligibility, patients discontinued all DMARDs four weeks before receiving the first injection of the study drug. Participants using over the counter preparations, NSAIDs, or oral corticosteroids (maximum daily dose equivalent of 10 mg of oral prednisone9,10) before screening were allowed to continue using them at pre-enrolment doses. The use of analgesics such as propoxyphene, codeine, acetaminophen plus codeine, or aspirin for pain relief was permitted.
Patients were randomised 1:1:1:1 to receive weekly sc injections of adalimumab (20, 40, or 80 mg) or placebo. Patients were instructed on proper self injection techniques and self administered the study drug. Off site self administration of the study drug continued throughout the study, with the exception of study visits, during which patients self administered adalimumab or placebo under supervision of study personnel.
After completing the 12 week study, patients entered a 40 week blinded continuation period, during which they were given the same weekly dose of adalimumab. Patients in the placebo group were switched to adalimumab 40 mg. Patients completing the 40 week period entered an open label continuation period, during which they received the same weekly doses of 40 mg adalimumab for another year. This report presents the results of the 12 week placebo controlled period. The results of the 40 week and 52 week continuation studies will be presented separately.
Patients with unbearable disease activity (a subjective condition determined by the patient and investigator) who completed at least eight weeks of the 12 week placebo controlled study were allowed to enter a rescue arm for the remainder of the study, during which they received increased doses of NSAIDs, corticosteroids, or other non-investigational agents; however, patients entering the rescue arm were not permitted to receive adalimumab until the end of the 12 weeks. Those who participated beyond week 12 were given open label adalimumab.
Patients
Patients 18 years of age or older were eligible for this study if they had a diagnosis of RA according to the revised 1987 American College of Rheumatology (ACR) criteria47 and active inflammatory synovitis, defined by a tender joint count (TJC) of ⩾12 and swollen joint count (SJC) of ⩾10 based on an examination of 68 and 66 assessed joints, respectively, and either an erythrocyte sedimentation rate (ESR) of ⩾28 mm/1st h or a serum C reactive protein (CRP) level ⩾20 mg/l. Only patients for whom treatment had failed with at least one traditional DMARD were eligible.
Exclusion criteria included joint surgery within two months before screening or an episode of infection requiring admission to hospital within 30 days before study entry. Treatment with either intra-articular or intramuscular corticosteroids within four weeks of prescreening or an investigational chemical or biological drug within two or six months, respectively, of prescreening were exclusionary criteria. Additionally, patients with impaired renal or hepatic function or an abnormal serum profile were excluded from the study. Patients’ body weight could not exceed 100 kg. Women of childbearing potential required a negative pregnancy test; the use of a reliable contraceptive method was mandatory.
Efficacy assessment
Clinical and laboratory efficacy assessments were conducted at screening, baseline, and two week intervals. ACR core criteria were monitored at each study visit: TJC and SJC; patient assessments of pain, disease activity, and disability (Disability Index of the Health Assessment Questionnaire (HAQ)48); doctor global assessment of disease activity; ESR; and CRP.49,50
The primary measure of efficacy was the number of patients meeting ACR20 response criteria at week 12. Patients were classified as responders if they fulfilled the ACR20 core criteria for a clinical response to treatment (that is, ⩾20% improvement in TJC and SJC plus ⩾20% improvement in three or more of the following five criteria: patient assessment of pain, patient global assessment of disease activity, doctor global assessment of disease activity, patient assessment of disability (Disability Index of the HAQ), and CRP).50
Secondary evaluations of efficacy included ACR50 and ACR70, defined in the same manner as ACR20 but with ⩾50% or ⩾70% degree of improvement, respectively. Other secondary outcomes were TJC, SJC, a battery of disease activity and health assessments, including the disease activity score 28 (DAS28), ESR, and CRP.49,50
Safety assessment
A complete physical examination was performed at study entry (screening), baseline, every two weeks, and, for those patients who withdrew, up to six months of follow up after the last study dose. Patients were monitored for vital signs and by electrocardiogram and laboratory tests, including haematology evaluations, serum chemistry (including a non-fasting lipid profile), coagulation parameters, and urine analysis. Additionally, serum was evaluated for the presence of antinuclear antibodies (ANAs), anti-double stranded DNA (anti-dsDNA) antibodies (if ANA levels were increased from baseline), and serum anti-adalimumab antibodies. ANA analysis was performed by an indirect immunofluorescence technique on HEp-2 cells. A positive ANA titre was defined as ⩾1/80. Anti-dsDNA positivity was established at >25 kU/l by an anti-DNA radioassay. Serum samples were analysed for anti-adalimumab antibodies using a validated double antigen, enzyme linked immunosorbent assay (ELISA). A positive anti-adalimumab signal was defined as a concentration exceeding 20 ng/ml that could not be suppressed by ⩾50% after the addition of 10% human serum to a sample. ANAs were measured at study entry, baseline, and monthly thereafter, and anti-adalimumab antibodies were measured at all scheduled study visits.
Data on adverse events (AEs), such as type, time of occurrence, time to resolution, severity, and causal relationship to treatment, were collected at the baseline visit and at each subsequent study visit. When appropriate, AEs and laboratory values were graded according to the Common Toxicity Criteria (CTC) of the National Cancer Institute (grade 1 = mild; grade 2 = moderate; grade 3 = severe; and grade 4 = life threatening or intractable). Laboratory AEs were automatically generated from CTC grade changes regardless of their clinical relevance. Additionally, AEs necessitating or prolonging hospitalisation, placing the patient at risk of death, or resulting in death, permanent or significant disability, congenital anomaly/birth defect, or other medically important conditions were categorised as serious AEs.
Statistical analysis
Assuming an ACR20 response rate of 20%, 90% power, and a drop out rate of 10%, a sample size of 64 patients in each treatment arm was required. Statistical significance was set at p⩽0.05 for all tests. The study was not powered to show a difference between treatment groups. Demographic and baseline characteristics were summarised by descriptive statistics, and treatment group comparability at baseline was determined using appropriate statistical tests. Efficacy analyses were performed for the intention to treat (ITT) population, defined as all randomised patients who received at least one double blind injection of study drug and for whom any assessment of efficacy under double blind conditions was available. One patient with Felty’s syndrome was excluded from the ITT population despite receiving a single injection of adalimumab 20 mg. No efficacy assessments under double blind conditions were available for this patient.
The response rates of the primary efficacy end point (ACR20 at week 12) for the adalimumab groups were compared with that of placebo using two sided Pearson’s χ2 tests, with α = 0.05. The closure principle was applied to take multiplicity of testing into account. The tests were performed sequentially versus placebo in order of descending adalimumab dose. Test results were considered to be significant if the p value of this test and the p values of all preceding tests were ⩽0.05. Patients not completing the 12 week trial (that is, those who were withdrawn or required rescue) despite fulfilling ACR criteria were considered non-responders. Primary and all secondary efficacy end points were summarised using descriptive statistics (that is, statistical characteristics, frequencies, percentages, confidence intervals). Descriptive treatment group comparisons between groups receiving adalimumab and placebo were performed at week 12 using two sided Pearson’s χ2 tests and analysis of covariance (ANCOVA). Within each treatment group, improvements from baseline in the seven ACR core criteria were evaluated at each time point for possible significant differences using the Student t test for paired observations.
All patients randomly allocated to receive adalimumab and who received at least one dose of adalimumab were included in the safety analyses. Vital signs and laboratory measures were described by statistical characteristics and by the frequency of abnormal values. AEs were analysed by frequency and reported as percentages of patients by randomised treatment.
RESULTS
Demographic and baseline characteristics
The demographic and baseline characteristics of the study population (table 1) were similar for all treatment groups. Patients ranged in age from 22 to 83 years, with an overall mean age of 52.4 years. Most patients were women (79%), and the majority were white (99%). The mean duration of RA was 10.0 years, and baseline TJC and SJC averaged 31.5 and 19.5, respectively. The baseline HAQ score was 1.71 and the baseline DAS28 was 7.0. CRP and ESR values (59 mg/l and 52.2 mm/1st h, respectively) substantially exceeded normal levels. Patient and doctor based assessments of disease activity and patient assessment of pain were 73.1, 64.9, and 71.7, respectively. Nearly 90% of patients reported one or more concomitant diseases. Before the study, all patients had used at least one DMARD (mean 3.75), with more than half (50.3%) having used from four to eight different DMARDs. The most commonly reported previously used DMARDs were MTX (92%), sulfasalazine (69%), parenteral gold (60%), chloroquine/hydroxychloroquine (48%), azathioprine (32%), penicillamine (30%), cyclosporin (29%), oral gold (17%), and cyclophosphamide (6%).
Demographic and baseline disease characteristics
Patient disposition
Of the 323 patients enrolled, 284 were randomly allocated to receive weekly sc injections of adalimumab 20 mg (n = 72), 40 mg (n = 70), or 80 mg (n = 72) or placebo (n = 70) (fig 1). The ITT population included all but the previously mentioned participant with Felty’s syndrome, who was withdrawn from the study after receiving one sc injection of adalimumab 20 mg. No efficacy assessments under double blind conditions were available for this patient. Two hundred and thirty three patients completed the study—186 (87%) patients in the adalimumab groups and 47 (67%) in the placebo group.
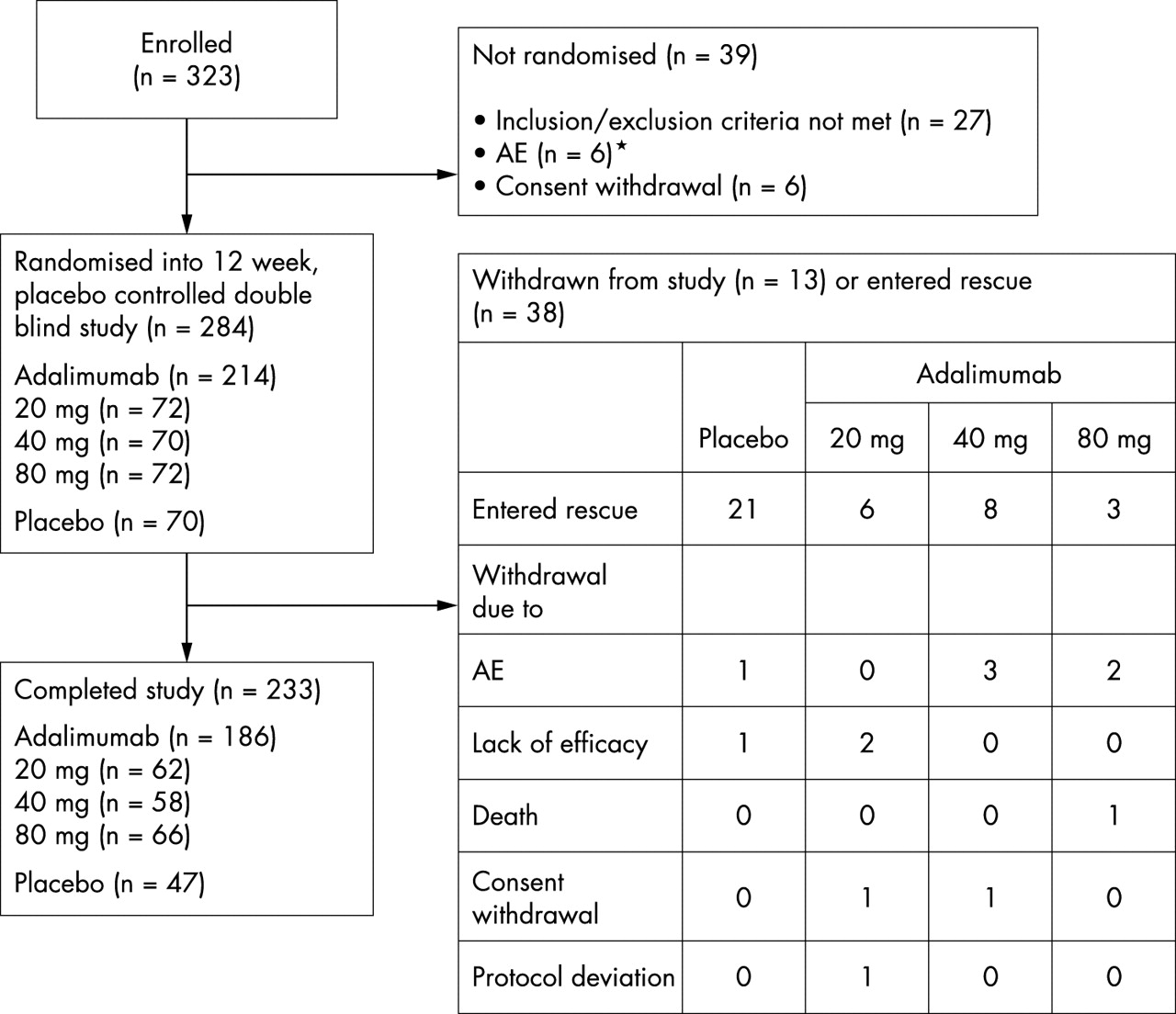
Disposition of patients.
Similar proportions of patients in the adalimumab and placebo groups were withdrawn during the study, 11/214 (5%) and 2/70 (3%) patients, respectively. From the adalimumab 20 mg group, 2 (3%) patients were withdrawn because of lack of efficacy and 1 (1%) patient because of protocol deviation and withdrawal of consent. From the adalimumab 40 mg group, 3 (4%) patients were withdrawn because of AEs (fever, raised ANA titres, gastrointestinal disorder) and 1 (1%) patient withdrew consent. The patient with raised ANA titres (of grade 3 severity and a maximum titre of 1/1280 at the time of withdrawal) was withdrawn because the investigator concluded that the condition was related to treatment with the study drug. Two (3%) patients in the adalimumab 80 mg group had AE related withdrawals (surgery, anxiety), and 1 (1%) patient in this group died from a myocardial infarction on day 86. Reasons for the 2 (3%) placebo withdrawals were AEs (vasculitis/paralysis/pain) and lack of efficacy. A greater proportion of patients in the placebo group (21, 30%) required rescue during the 12 week study period because of uncontrolled disease activity than those receiving adalimumab (17, 7.9%).
Efficacy assessments
ACR response
The ACR20 response was significant for each adalimumab group at each evaluation throughout the study relative to placebo (p⩽0.01 for all comparisons, two sided Pearson’s χ2 test) (table 2, fig 2A), with 71% (82/115) of the ultimate ACR20 response rate to adalimumab treatment being achieved at week 2. At week 12, 36 (51%), 40 (57%), and 39 (54%) patients treated with adalimumab 20, 40, and 80 mg, respectively, were ACR20 responders, compared with 7 (10%) placebo patients. Maximal ACR20 response rates for the adalimumab 20, 40, and 80 mg groups were reached at week 12 (36, 51%), week 10 (44, 63%), and weeks 6 and 12 (39, 54%), respectively.
American College of Rheumatology (ACR) response to treatment at weeks 2 and 12. Results are shown as number (%)

Time course of American College of Rheumatology responses. (A) ACR20 response; (B) ACR50 response; (C) ACR70 response. Student’s paired t test for paired observations: *p = 0.01 v baseline; †p = 0.01 v baseline; ‡p = 0.05 vv baseline.
ACR50 response rates for the adalimumab 40 and 80 mg groups were statistically better than those for placebo (p⩽0.05) at any given study visit, and for the 20 mg group at week 4 and all subsequent study visits. Fifty eight percent (29/50) of the ultimate ACR50 response rate was achieved at week 4. At week 12, 17 (24%), 19 (27%), and 14 (19%) patients in the adalimumab 20, 40, and 80 mg groups, respectively, had an ACR50 response, compared with 1 (1%) patient in the placebo group (p⩽0.001 for all comparisons) (table 2, fig 2B). Maximal ACR50 response for the adalimumab groups (by ascending dose) was attained by week 12 (17, 24%), week 10 (20, 29%), and week 8 (14, 19%), respectively.
ACR70 responses at week 6 reached significance versus placebo for the two lower doses of adalimumab and at week 10 for the highest dose. At week 12, all adalimumab doses were associated with statistically significant ACR70 response relative to placebo: 8 (11%), 7 (10%), and 6 (8%) patients in the 20, 40, and 80 mg groups, respectively (p⩽0.01; p⩽0.01; p⩽0.05, respectively) (table 2, fig 2C). No patients receiving placebo attained an ACR70 response. Maximal responses for the treatment groups receiving adalimumab 20, 40, and 80 mg were reached at week 12 (8, 11%), week 10 (9, 13%), and week 12 (6, 8%), respectively.
ACR responders in all adalimumab groups achieved ACR response status more rapidly than the placebo responders (table 3). The mean times to the onset of ACR responses were dose dependent, with the adalimumab 20 and 40 mg groups having similar mean times to the onset of ACR20 response and the 80 mg group having a somewhat shorter time. The mean times to meeting ACR50 and ACR70 criteria also were dose dependent.
Mean time (weeks) to achieving response according to the American College of Rheumatology (ACR)
ACR core criteria
Treatment with all doses of adalimumab resulted in rapid, significantly improved signs and symptoms of RA relative to placebo, as assessed by improvements in ACR core criteria (p⩽0.01 v placebo for all comparisons, ANCOVA) (table 4).
American College of Rheumatology (ACR) core response criteria: baseline values and mean absolute changes from baseline to weeks 2 and 12*
Patients experienced an immediate, rapid decline in mean TJC after the initial injection of adalimumab. At week 2, mean reductions of 7.7, 9.5, and 11.4 were reported for the adalimumab 20, 40, and 80 mg groups, respectively; the placebo group showed little improvement (that is, a mean decrease of 1.0). All adalimumab groups exhibited significant improvement in TJC at each subsequent evaluation, with actual reductions of 14.0, 15.3, and 15.2, respectively, at week 12, representing mean changes of 44.2%, 49.4%, and 46.8%, respectively (p⩽0.001 v placebo for all comparisons).
A similar degree of rapid clinical improvement was shown by the immediate decline in the mean number of swollen joints in patients receiving adalimumab. By week 2, all adalimumab groups attained significant mean reductions in SJC from baseline—4.6, 5.0, and 5.4 for adalimumab 20, 40, and 80 mg, respectively, compared with 0.9 for placebo. Actual decreases in SJC at week 12 averaged 8.1, 9.6, and 10.7, respectively, representing improvements of 41.3%, 51.3%, and 54.6%, respectively (p⩽0.001 v placebo for all comparisons). Patients receiving placebo showed little improvement in SJC at week 12 (mean decrease of 2.8, 13.9%).
HAQ Disability Index scores for adalimumab decreased significantly from the first evaluation to the end of the study. Decreases in HAQ scores at week 2 averaged 0.34, 0.28, and 0.30 for adalimumab 20, 40, and 80 mg, respectively, compared with an increase of +0.05 for placebo, indicating worsening of disability. Mean HAQ Disability Index scores continued to decrease for all adalimumab groups up to week 12, with reductions of 0.45, 0.47, and 0.48, respectively (p⩽0.001 v placebo for all comparisons) and 0.04 for placebo.
At each assessment, significantly lowered CRP concentrations were reported for all adalimumab groups, with reductions at week 12 reaching 24, 32, and 34 mg/l for adalimumab 20, 40, and 80 mg, respectively (p⩽0.001 v placebo for all comparisons) and little change for placebo. Additionally, significant reductions in mean ESR from baseline occurred at week 2 (p⩽0.001 v placebo for all comparisons). By week 12, ESR values had decreased 14.2, 17.9, and 17.3 mm/1st h for adalimumab treatment groups by ascending doses (p⩽0.01, p⩽0.001, p⩽0.001 v placebo for adalimumab 20, 40, and 80 mg, respectively) and by 2.0 mm/1st h for placebo.
Patient and doctor global assessments of disease activity and patient assessment of pain for all adalimumab groups significantly improved at each study visit (p⩽0.001 v placebo for all comparisons).
Laboratory results
Clinically significant improvements occurred in several haematological laboratory values in adalimumab patients, including increased haemoglobin, packed cell volume, and percentages of lymphocytes as well as decreased platelet counts, fibrinogen levels, and percentages of neutrophils (data not shown).
Safety assessments
Table 5 presents an overview of AEs, including those that occurred most frequently (⩾10%) by treatment group. The most commonly reported treatment related AE was laboratory test abnormality, particularly decreased lymphocytes, which occurred with marked difference (⩾10% more frequently) in patients randomised to placebo (39, 56%) compared with those randomised to adalimumab (97, 45%). Two AEs occurred in adalimumab patients with 10% or greater frequency than with placebo. Reactions occurring at the injection site—erythema and/or itching, haemorrhage, swelling, or pain—were reported in 21 (29%), 16 (23%), and 21 (29%) patients receiving adalimumab 20, 40, and 80 mg, respectively, versus 4 (6%) patients randomised to placebo (p⩽0.01 v placebo for all comparisons). Hyperlipidaemia (triglycerides >2.26 mmol/l was reported in 18 (25%), 22 (31%), and 22 (31%) patients receiving adalimumab 20, 40, and 80 mg, respectively, versus 13 (19%) patients receiving placebo. None of the commonly reported treatment emergent AEs showed a dose-response relationship to adalimumab.
Overview of adverse events (AEs). Results are shown as the number (%) of patients
The occurrence of serious AEs with adalimumab did not exceed that with placebo—16 (7%) versus 7 (10%), respectively. Serious infections occurred in 4 (2%) patients in the adalimumab dose groups. In the adalimumab 40 mg group, 1 (1%) patient had a gastrointestinal disorder of an infectious nature and 1 (1%) had mild bronchitis. Two (3%) patients receiving adalimumab 80 mg developed serious infections, one a urinary tract infection and the other a bacterial infection with fever. Both infections were deemed by investigators as possibly related to adalimumab treatment, and the patients were withdrawn from the study, each requiring additional drugs and admission to hospital. (The source of the bacterial infection was never identified and the fever subsided after five days of antibiotic treatment.) All serious infections resolved or were resolving by week 12. A fatal myocardial infarction occurred on day 86 of the study in a 65 year old man with pre-existing cardiac disease who was randomised to adalimumab 80 mg and was receiving a concomitant antihypertensive drug. The investigator judged the death unlikely to be related to the study drug. No drug related toxicity was indicated by haematology, coagulation, clinical chemistry, or urine analysis evaluations.
During the study, changes in ANA status—from baseline negative to positive and from baseline positive to negative—were seen commonly at all evaluations. Among the 283 patients (adalimumab = 213, placebo = 70) evaluated for ANA status during the study, 37/71 (52%), 29/70 (41%), and 34/72 (47%) patients in the 20 mg, 40 mg, and 80 mg adalimumab groups, respectively, had ANA positivity (titres ⩾1/80) compared with 26/70 (37%) patients in the placebo group. Of the adalimumab treated patients testing positive to ANAs (n = 100), 3 (3%) patients also tested positive to anti-dsDNA antibodies (that is, >25 kU/l): 1/37 (3%) and 2/34 (6%) patients in the 20 and 80 mg groups, respectively. No patients in the placebo group tested positive to anti-dsDNA antibodies. No patients developed lupus-like syndrome, and no malignancies were reported. During the study, anti-adalimumab antibodies were detected in the sera of four patients.
DISCUSSION
Adalimumab is the first fully human monoclonal antibody to TNFα to enter clinical trials for the treatment of RA. Early phase I/II trial results with adalimumab monotreatment have been very encouraging.17–18,34,37,39
The current study was designed to evaluate the effectiveness, dose response, safety, and tolerability of weekly sc injections of various doses of adalimumab under placebo controlled conditions. Study participants generally had severe, recalcitrant, long term RA. Despite prior treatment with an average of nearly four traditional DMARDs, these patients continued to exhibit persistent disease activity, including large numbers of tender and swollen joints, thus placing them at risk of further joint destruction and worsening disability.
Patients receiving adalimumab experienced significantly better improvement in their physical condition, evidenced by the statistically significant ACR20 and ACR50 responses at virtually all study assessments for each adalimumab group compared with placebo. At the 12 week assessment, more than 50% of patients in each adalimumab group were ACR20 responders and about 20% had attained an ACR50 response. Additionally, at week 12, all doses of adalimumab produced a statistically significant ACR70 response versus placebo.
Adalimumab is associated with rapid clinical response rates. In one study, clinically relevant responses were reported to occur at 24 hours to one week after a single intravenous injection of adalimumab.34 In the current study, sc injections of adalimumab resulted in significantly better results by the first study assessment at week 2, with the majority of eventual responders already fulfilling ACR20 or ACR50 response criteria, compared with 8% of eventual responders receiving placebo. The mean times to the onset of ACR20 and ACR50 responses were consistently shorter for all adalimumab doses than for placebo.
The rapid clinical response to adalimumab treatment also was shown by the early (that is, week 2) statistically significant decline in the number of swollen and tender joints as well as improvements in other ACR core criteria compared with placebo in the DMARD refractory population. All improvements were immediate and sustained. More than half of the progress in disease activity as reflected in the ACR core criteria occurred within two weeks of the initial dose of adalimumab. At all subsequent evaluations, all doses of adalimumab were associated with statistically better improvements in the ACR core criteria than placebo. Patient assessed outcomes such as functional status (HAQ), RA related pain, and global disease activity significantly improved over the course of treatment with adalimumab. Here again, enhanced physical functioning, as reflected by these measures of disease activity, was rapid, with more than half of the total improvement in scores occurring within the first two weeks of treatment.
It is also interesting to note that at week 2, adalimumab treated patient assessments of disease activity, including pain and physical functioning, were consistently better than doctor assessed disease activity, even though at baseline, patients assessed their disease state as being more severe than did the investigators (73.1 v 64.9, respectively, on the 100 mm visual analogue scale).
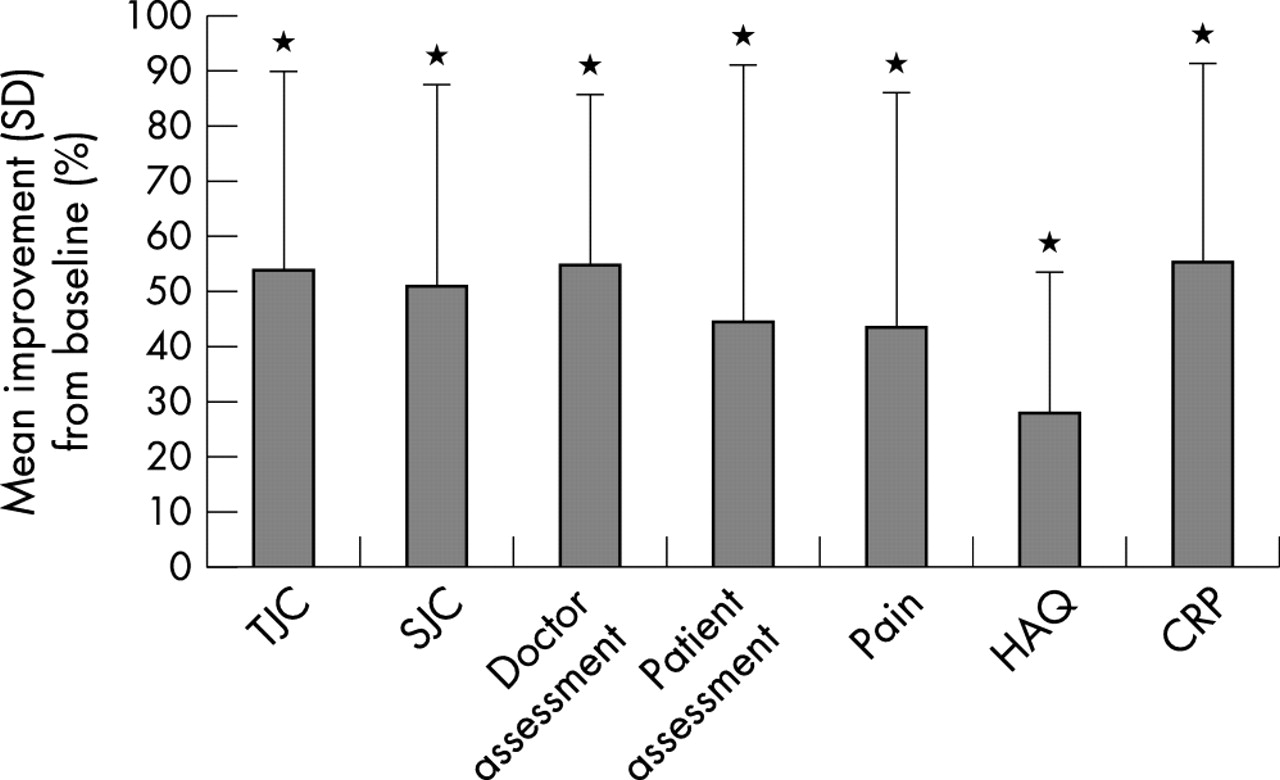
In most measures of efficacy at week 12, adalimumab 40 mg was associated with better results than the other doses. Figure 3 illustrates the improvements in ACR criteria attained by this group over the entire treatment period. However, it is important to note that the study was neither designed nor powered to show statistical differences between the adalimumab groups. Compared with placebo, all adalimumab groups had consistently better ACR responses and improvements in ACR core criteria from baseline.

{kind=link}
{kind=link}
{kind=link}
Improvement in American College of Rheumatology core criteria at 12 weeks with adalimumab 40 mg. Student’s t test for paired observations: p = 0.001 v baseline.
Overall, adalimumab was safe and well tolerated at all doses. AEs commonly occurred in all groups, a finding consistent with a study group having longstanding active RA. It should be noted that at baseline, more than half of the patients had AEs attributable to the disease state or to concomitant drugs. In general, treatment emergent AEs were mild or moderate in intensity, and those of a more serious nature resolved or were resolving by the end of the study. A 65 year old man taking adalimumab 80 mg died of a myocardial infarction on day 86 of the study. This patient, who had pre-existing cardiac disease and cardiovascular risk factors, was at increased risk of myocardial infarction and was treated with the antihypertensive amlodipine. The patient’s total cholesterol level was slightly raised (5.85 mmol/l) at week 12 but was below baseline level (7.40 mmol/l). The investigator judged the relationship of the death to the study drug as being unlikely.
The high incidence of laboratory abnormalities—nearly 50% overall—can be attributed to the automatic reporting of these events whenever changes in CTC grade occurred (which may have been small departures from normal ranges), regardless of their clinical relevance.
Reactions at the site of the sc injection are commonly associated with sc administered drugs used to treat RA. Clinical trials with other sc administered biological agents in patients with RA reported injection site reactions of 37% (etanercept)28 and more than 70% (anakinra).51 In the current study, injection site reactions occurred with more statistically significant frequency among adalimumab treated patients than placebo treated patients (27% v 6%; p⩽0.01 v placebo); but, overall, these reactions were mild and manageable. Proteinuria also occurred more frequently in the adalimumab treated patients (7% v 0%; p⩽0.05 v placebo); however, no systematic or dose related trends were seen.
Adalimumab treated patients demonstrated mild increases in serum lipid levels in comparison with placebo treated patients. It is difficult to analyse such data because patients in both groups had blood samples drawn while not fasting, and the two groups were not matched for dietary intake and baseline lipid profiles. However, in general, patients with RA, when untreated, demonstrate altered lipoprotein profiles (that is, low cholesterol concentrations in all lipoprotein fractions and low triglyceride concentrations in the very low density and high density lipoprotein fractions).52 Treatments that reduce inflammation generally effect normalisation in these profiles.52 It has been suggested that altered lipid patterns with untreated active RA may be associated with an increased risk of atherosclerosis53; therefore, effectively controlling the inflammatory state in RA may have additional benefits.54
Because the mechanism of action of cytokine treatments such as adalimumab is inhibition of an immunologically active cytokine, these agents can be associated with an increased incidence of infectious complications. During this study, patients were carefully monitored for episodes of serious infection and immunological reactions. The incidence of serious infections in adalimumab treated patients (2%) was slightly less than the expected incidence of these events in patients with RA receiving immunosuppressant drugs (2.9%).55 No cases of lupus-like syndrome were reported among patients with an increase in autoantibody production. Four patients developed anti-adalimumab antibodies during the study. Because adalimumab has sequences that are germline human in the framework regions, it is expected to have reduced immunogenicity compared with antibodies that contain sequences derived from non-human species. Nevertheless, low level anti-idiotypic responses would be expected based on the theory that human antibodies may be part of an idiotypic network as described by Jerne.56
Because of the small number of cases, no overall conclusions can be drawn about the overall efficacy and safety of patients who develop anti-adalimumab antibodies.
The three adalimumab doses were similarly tolerated, with no dose limiting effects between groups in relation to any AE. The high patient retention rate (87%) among DMARD refractory patients receiving adalimumab (v 67% for placebo) indicated that study participants were willing to cope with occasional side effects as well as with self administration of the study drug to sustain improved disease control.
In conclusion, adalimumab treatment significantly altered the course of disease in patients who had considerable disease activity of long duration and who had not adequately responded to traditional DMARDs. All adalimumab doses acted more rapidly and were more efficacious than placebo, with the 80 mg dose of adalimumab not providing any additional benefit over the 40 mg dose. Treatment related reductions in disability plus marked improvements in acute phase reactants such as CRP and ESR suggest that adalimumab treatment may halt disease progression. Recent reports substantiate this outcome among patients receiving adalimumab monotreatment over a one and two year period.35,36 Nevertheless, these results need to be confirmed in large radiographic studies. Adalimumab treatment was safe and convenient, allowing patients to self administer the sc injections at home. No systemic allergic reactions were reported with adalimumab treatment, enabling long term administration without the concurrent use of immunosuppressant drugs such as MTX.
Patients completing this 12 week placebo controlled study were subsequently entered into a 40 week blinded continuation study of adalimumab, followed by an open label study for an additional year in order to validate the chronic efficacy and safety of adalimumab. The results of these continuation studies will be presented separately.
Acknowledgments
The authors gratefully acknowledge the study group that conducted this phase II trial: Austria: Dr B Leeb, Dr M Kögel, Dr W Ebner, Krankenhaus der Stadt Wien-Lainz, Vienna; Dr K Machold, Dr G Eberl, Dr M Köller, Allg Krankenhaus der Stadt Wien, Vienna. Belgium: Professor Dr M Malaise, Dr O Kaye, Dr B André, CHU Liège, Liège; Professor Dr E M J C Veys, Dr K Vanderkerckhove, University Hospital Ghent, Ghent. France: Professor Dr M Dougados, Dr M Nguyen, Hôpital Cochin, Paris; Professor Dr O Meyer, Dr M de Bandt, Hôpital Bichat, Paris; Professor D J Sany, Dr V Ferrazzi, Hôpital Lapeyronie, Montpellier; Professor Dr J Sibilia, Dr F X Limbach, Hôpital de Hautepierre, Strasbourg. Germany: Professor Dr G R Burmester, Dr U Alpermann, Dr S Scholze, Universitätsklinikum Charite Humboldt-Universität, Berlin; Professor Dr J R Kalden, Dr C Antoni, Dr C Dechant, Medizinische Klinik III mit Poliklinik der Friedrich-Alexander Universität, Erlangen; Professor Dr H H Peter, Dr E Sandrock, Dr B Bannert, Klinikum der Albert-Ludwigs-Universität, Freiburg; Professor D R Rau, Dr S Simianer, Dr G Herborn, Evangelisches Fachkrankenhaus, Ratingen; Professor Dr M Schattenkirchner, Professor Dr K Krüger, Dr J Wastlhuber, Universität München, Munich; Professor Dr H Zeidler, Dr J Hülsemann, Dr E Riechers, Medizinische Hochschule Hannover, Hannover. Greece: Professor Dr H Moutsopoulos, Dr F N Skopouli, Dr G Linardaki, University of Athens School of Medicine, Athens. The Netherlands: Professor Dr F C Breedveld, Dr C F Allaart, Academisch Ziekenhuis Leiden, Leiden; Professor Dr L B A van de Putte, Professor Dr P L C M van Riel, Dr A A den Broeder, Academisch Ziekenhuis Nijmegen, Nijmegen. Spain: Dr J A Jover, Dr C Hernandez-Garcia, Hospital Universitario San Carlos, Madrid; Dr J Gomez-Reino, Hospital Xeral de Galicia, Santiago-Compostela. Switzerland: Professor Dr B Michel, Dr T Langenegger, Dr D Frey, Universitätsspital-Rheumaklinik, Zurich; Professor Dr T Vischer, Dr N Buchs, Hôpital Beau-Sejour, Geneva. United Kingdom: Professor Dr P Bacon, Dr E Rankin, University of Birmingham, Birmingham; Professor Dr P Emery, Dr K Ahmed, Old Home Leeds General Infirmary, Leeds; Professor Dr D Isenberg, Dr S Hussain, University College London Hospitals, London; Dr D Blake, Dr D Speden, The Royal National Hospital for Rheumatic Diseases, Avon Bath.
This study was supported by Abbott Laboratories.
REFERENCES
Footnotes
-
Acting editor for this paper was Professor Frank Wollheim.