Article Text

Abstract
Biological therapies directed at proinflammatory cytokines have irrevocably changed the landscape of treatment of rheumatoid arthritis (RA) and other autoimmune diseases. With the advances in our knowledge in cytokine signalling, the question emerges whether targeting intracellular signalling might also be a safe and efficacious strategy. Janus kinases or Jaks are critical for a large family of cytokines and the first Jak inhibitors has been approved by the FDA. It is therefore timely to consider this new category of drugs and reflect on their potential roles, present and future, in the treatment of RA and related disorders.
- Rheumatoid Arthritis
- Autoimmune Diseases
- Treatment
Statistics from Altmetric.com
Role of type I/II cytokines in rheumatoid arthritis and related diseases
Cytokines are critical for host defence and immunoregulation, but also major players in the immunopathogenesis of autoimmune diseases. Practically, rheumatologists can adduce the success of recombinant cytokine receptors and monoclonal antibodies against cytokines as evidence for the immunopathological role of these factors1 What the practising physician may be less cognisant of is the complexity of cytokines and the diversity of their structure.
Based on structure, several major families of cytokines can be recognised. Two major classes are the so-called type I and type II cytokine receptors. Type I receptors bind several interleukins (ILs), colony stimulating factors and hormones such erythropoietin, prolactin and growth hormone. Type II receptors bind interferons and IL-10 related cytokines.
Genome-wide association scans have identified a plethora of single-nucleotide polymorphisms (SNPs) conferring genetic susceptibility in autoimmune diseases such as rheumatoid arthritis (RA),2 psoriasis,3 inflammatory bowel disease (IBD)4 and ankylosing spondylitis.5 Polymorphisms of genes encoding type I cytokine receptors and their signalling elements are now firmly linked to various autoimmune diseases. For instance, IL-23R, IL12B, JAK2 and STAT3 polymorphisms are associated with IBD and psoriasis. STAT4 polymorphisms are associated with RA, systemic lupus erythematosus and Sjogren syndrome. Other evidence of culpability of type I/II cytokines in autoimmunity comes from their detection in the context of disease. RA, for instance, is associated with overproduction of IL-6, IL-12, IL-15, IL-23, granulocyte-macrophage colony stimulating factor (GM-CSF) and interferons.2
Signalling via type I/II cytokine receptors
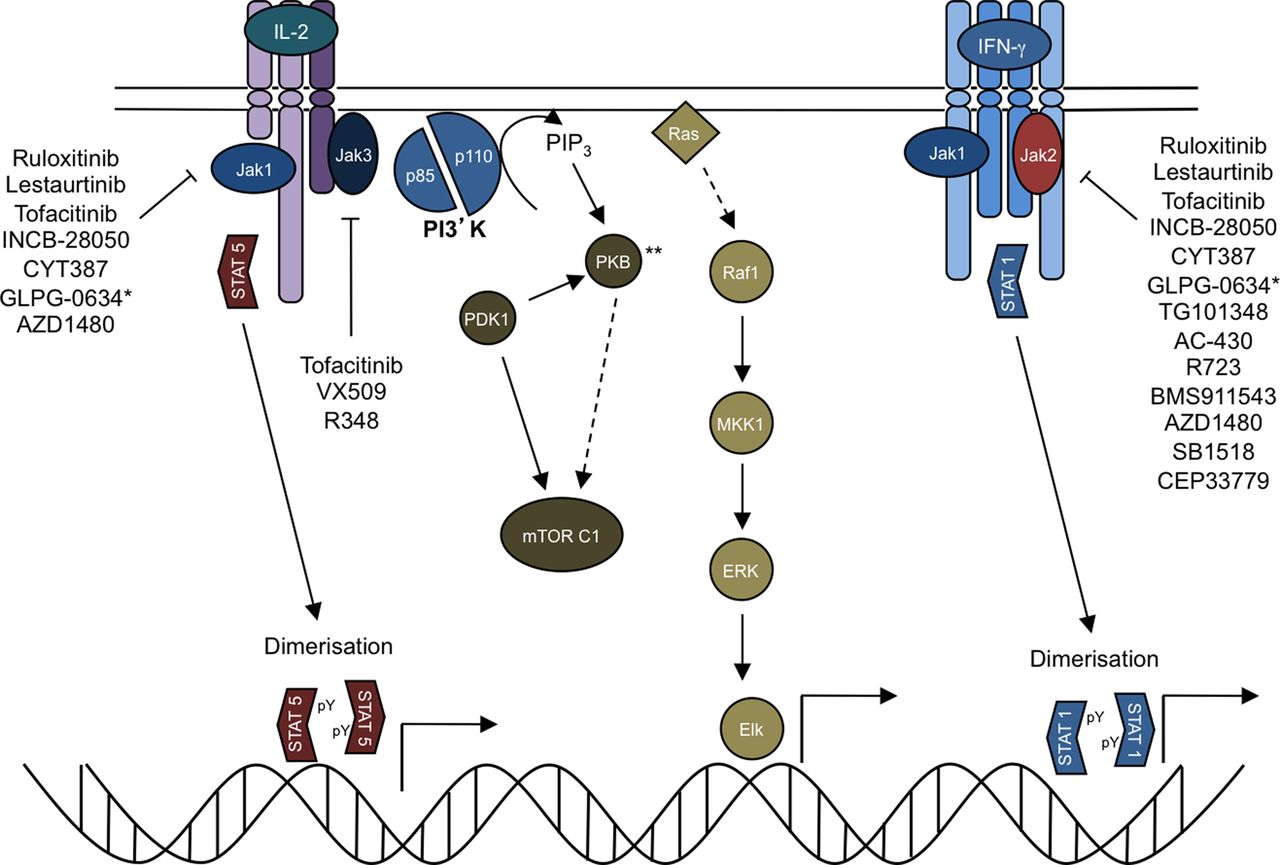
In contrast to other receptors, whose intracellular domains encode kinase or other enzymatically active domains, these receptors lack such elements. Instead, the cytoplasmic domain of type I and II cytokine receptors binds to members of a specific kinase family, known as the Janus kinases (Jaks) which include Tyk2, Jak1, Jak2 and Jak3 (figure 1).6 Cytokine receptors are paired with different Jaks, which are activated on cytokine binding (figure 2). Because Jaks are phosphotransferases, they catalyse the transfer of phosphate from ATP to various substrates such as cytokine receptors. This modification allows the recruitment of various signalling molecules including members of the signal transducer and activator of transcription (STAT) family of DNA binding proteins.7 STATs are another important Jak substrate. Phosphorylation of STATs promotes their nuclear accumulation and regulation of gene expression.

Usage of different Janus kinases (Jaks) by various cytokines.
{kind=link}
{kind=link}
Janus kinase (Jak) inhibitors (Jakinibs) block multiple aspects of cytokine signalling. Cytokine binding to its cognate receptor leads to phosphorylation of the intracellular domain of the tyrosine kinase receptor by specific Jaks. Signal transducer and activator of transcriptions (STATs) are then recruited, bind to the receptor and become phosphorylated by Jaks. This results in STAT dimerisation, translocation and regulation of gene transcription. Cytokines also activate the protein kinase B (PK; also known as Akt, which is named after the Ak mouse strain that predisposes to thymoma) and mammalian target of rapamycin (mTOR). Though not carefully studied, it is highly likely that blocking proximal cytokine signals will disrupt all downstream pathways. ** Also referred to as AKT.
Elegant work from mutagenised cell lines and later, knockout mice, supports the critical and specific role of Jaks signalling by type I/II cytokines and not other pathways.8 In vivo evidence of the non-redundant functions in humans emerged from primary immunodeficiency patients.9
It is important both conceptually and practically to bear in mind that receptors for cytokines like tumour necrosis factor (TNF), IL-1 and IL-17 are structurally distinct from type I/II cytokine receptors; these cytokines are not dependent upon Jaks for signalling.10–12
Targeting kinases
Work over the past 25 years has established that protein phosphorylation is a fundamentally important mode of intracellular signal transduction.13 Thanks to the completion of the human genome, we now know the identity of all these players: there are over 500 kinases in the human kinome, which can be divided into eight families. The Jaks belong to the tyrosine protein kinase family of which there are 90 members. Structurally, the catalytic domains of all these kinases are highly conserved. Consequently, one might imagine that generating therapeutically useful kinase inhibitors would be an enormous challenge. However, it is now clear that kinases are actually very good targets and chemists have become skilled in generating reasonably selective inhibitors. So far, 13 inhibitors have entered clinical use and are approved by the FDA. Clearly, the overall strategy of targeting kinases is no longer theoretical.
Jakinibs in 2013
The critical function of Jaks in cytokine signalling has made them targets for industry to consider. At present there are a number of Jak inhibitors (Jakinibs) in clinical use or being tested in clinical trials.
Ruxolitinib and baracitinib
The discovery that gain-of-function JAK2 mutations underlie the myeloproliferative disorders including polycythaemia vera, essential thrombocythemia and myelofibrosis (MF) was a great breakthrough in understanding the pathophysiology of these disorders.14 The identification of these mutations also provided a rationale for purposefully targeting this enzyme. Ruxolitinib is a Jak1/2 inhibitor that is now approved by the FDA for the treatment of intermediate- and high-risk MF.15–17 Ruxolitinib reduces splenomegaly and systemic symptoms and also improves overall survival.
However, ruxolitinib has also been studied in RA where preliminary results were promising in terms of efficacy and safety in a phase IIa trial.18 Ruxolitinib has also been used as a topical formulation in psoriasis with promising results.19 Like ruxolitinib, baracitinib (formerly designated INCB028050) is also a Jak1/Jak2 inhibitor which showed efficacy in a highly active RA patient group resistant to disease modifying drugs and biological, with superior results in higher doses up to 4 or 8 mg once daily within 2 weeks; dose dependent side effects included decrease of haemoglobin and neutrophil count and increase of low density lipoprotein (LDL) and creatinine, but there was good overall tolerability.20
Tofacitinib
Tofacitinib (formerly CP-690550) was actually the first Jak inhibitor to be tested in the clinic. Tofacitinib inhibits Jak3 and Jak1 and to a lesser extent Jak2. It has little effect on Tyk2.21 Across the kinome it has selectivity, remarkably sparing other kinases, showing its high specificity compared to others.22
Because of the prominent role of type I/II cytokines in driving autoimmunity and the effect of tofacitinib on these cytokines, this drug has been tested in a range of settings from RA, IBD and psoriasis to renal transplantation rejection and dry eyes.23–28 Phase III trials have shown efficacy for tofacitinib in RA patients who have failed disease-modifying anti-rheumatic drugs (DMARDs), both as monotherapy24 and in combination with methotrexate.25 These findings are consistent with prior phase II trials.27 ,29 Of interest, tofacitinib was not inferior to standard of care therapy, namely, adalimumab in combination with methotrexate30 There is evidence that structural damage was also averted,31 however, further investigation will be needed to substantiate this. Of note, tofacitinib was efficacious in patients who failed with multiple biologicals.24 For all these reasons, tofacitinib has recently been approved by FDA in the USA for moderate to severe RA in patients with inadequate responses to methotrexate.
Other Jakinibs
The picture is made complicated in that VX-509, a reportedly specific Jak3 inhibitor, was also efficacious in a phase IIa study in RA.32 Moreover, a reportedly selective Jak1 inhibitor, GLPG0634, also met its primary endpoint in a phase IIa RA trial with no anaemia and no lipid abnormalities observed.33 CEP-33779, a selective Jak2 inhibitor, showed efficacy in two preclinical arthritis models.34 Thus, the relative contribution of the different Jaks in disease pathogenesis and the utility of selective blockade remains to be determined. At present, there are no selective Tyk2 inhibitors in clinical trials.
Mechanism of action of first-generation Jakinibs
An increasing body of evidence implicates specific cytokines and cell subsets as drivers of pathogenesis in different autoimmune diseases. Many of these key cytokines use the Jak/STAT pathway to exert their effects, rendering them amenable to therapeutic blockade with Jakinibs. Given the apparent pathogenic role of a variety of cytokines like IL-6, IL-12, IL-23, interferons and GM-CSF in RA, psoriasis, IBD, AS and other autoimmune diseases, the ability of Jakinibs to block such cytokines is likely a major aspect of their mechanism of action.
Mechanistically, tofacitinib blocks common γc cytokines including IL-2, IL-4, IL-7, IL-9, IL-15 and IL-21, all of which are signal through Jak3. In addition, it blocks Jak1, which would result in inhibition of the gp130 family including IL-6 and IL-11, as well as the type II cytokine receptor family such as interferon (IFN)-α/β, IFN-γ and IL-10. To a lesser extent the drug blocks Jak2 and therefore blocks the βc family such as IL-3, IL-5 and GM-CSF as well as EPO (erythropoietin) and IFN-γ.6 Because tofacitinib blocks Jak1 and Jak2, it interferes with the differentiation of IFN-γ producing Th1 cells. It also blocks the generation of pathogenic Th17 cells, which are dependent on IL-23.21 ,35 Because tofacitinib blocks IL-4 and IL-21, it might be anticipated that it will interfere with the function of B cells and follicular helper T cells. In addition to blocking the function of lymphocytes (adaptive immunity), tofacitinib also blocks innate immune responses. Specifically, tofacitinib blocks the effects IL-6 and interferons and thereby inhibits chemokine production from synovial fibroblasts.35 ,36 In a sepsis model, which is dependent on IFN-γ, tofacitinib blocked the production of TNF and IL-1.21 Thus, tofacitinib can interfere with the production and action of TNF. However, TNF signalling per se is not affected; rather, tofacitinib blocks autocrine effects of interferons that mediate TNF effects.36 In patients with RA treated with tofacitinib, serum levels of IL-6 were significantly decreased; presumably, this is due to effects on type I/II cytokines that induce IL-6.37 In an RA animal model, tofacitinib abrogated osteoclast-mediated arthritic joint structural damage by decreasing receptor activator of nuclear factor kappa-B ligand (RANKL) production.38
Because ruxolitinib and baracitinib inhibit Jak1 and Jak2, they block many of the same cytokines as tofacitinib. Deletion of Jak3 impedes lymphocyte development because of its requisite role in cγc cytokine signalling.39–41 However, gene targeting of Jak1 also results in a severe combined immunodeficient phenotype.42 From this perspective, the expectation would be that these drugs might have very similar mechanisms of action, in terms of the cytokines that are blocked (figure 2).
Side effects of Jakinibs
An important side effect of Jakinibs is serious bacterial, mycobacterial, fungal and viral infections. In the phase II, III and long extension trials of tofacitinib among opportunistic infections, tuberculosis (TB) was reported in 12 cases, 11 of which were initially negative on screening for TB, and 10 occurred in patients from endemic countries. Increased frequency of non-disseminated herpes zoster was also reported which may reflect reduction of NK cells by virtue of Jak1 or Jak3 blockade. Whether this accounts for viral infection susceptibility remains to be established. Longer duration adequately powered trials are needed to estimate the risk of common and opportunistic infections. A potential advantage of Jakinibs compared to biologicals with respect to infection risk is the relatively short half-life of the former; if infections occur, the drug can be stopped and the immunomodulatory effect is transient.
Jakinibs can cause anaemia, thrombocytopenia and neutropenia, likely related to Jak2 inhibition, which is important for EPO signalling and the actions of colony stimulating factors. When used for treatment of MF in the setting of thrombocytopenia, the dose of ruxolitinib needs to be adjusted accordingly.
Use of Jakinibs is associated with hypercholesterolaemia. However, this is also consistently observed in RA trials with tocilizumab, implying that high LDL, triglycerides and high density lipoprotein may be mediated by blockade of IL-6 signalling. Standard anti-hyperlipidaemic therapy improves the metabolic profile but the overall risk for cardiovascular morbidity will need to be determined in the long term.43
Small increases in creatinine have been observed with tofacitinib; it is unclear if these effects are related to the drug's mechanism of action.
A concern regarding chronic treatment with Jakinibs pertains to the possibility of increased cancer risk. Interferons and NK cells are important in tumour surveillance and the blockade of their action provides the theoretical rationale for development of malignancies mandating increased clinical vigilance.44 The rate of lymphomas or other lymphoproliferative disorders in phase III and long extension studies of tofacitinib in RA was 0.07 per 100 patient-years (95% CI 0.03 to 0.15) which is comparable with studies of other biologicals and the general RA population.25
Overall, the use of Jakinibs in clinical practice depends on the efficacy and safety ratio compared to standard of care therapy in a carefully selected patient population.
The future of Jakinibs in treating autoimmune disease
Clinical use of Jakinibs
Over the past decade, the biologicals have clearly raised the bar with respect to treatment of rheumatic disease. They are highly effective and remarkably safe. However, not all patients respond. Exactly how Jakinibs will fit within the rheumatologist's armamentarium remains to be seen. It will be of interest to see how a new, highly effective oral agent will be embraced relative to established parenteral drugs. An exciting development is that patients who fail with biologicals respond to Jakinibs.
Jakinibs in other diseases
Trials in psoriasis, IBD and transplantation are presently ongoing. In preclinical studies, Jakinibs appear to have efficacy in lupus models.34 ,45 ,46 The possibility of treating patients with systemic lupus erythematosus is attractive given the prominence of the ‘interferon signature’ in this disease.47–49 Asthma and allergy is associated with Th2 responses and the action of IL-4. Jak1 and Jak2 are important for IL-4 signalling and the potential utility of tofacitinib and other Jakinibs in these disorders is supported by preclinical data.50
Selective versus pan-Jak inhibitors
The kinome is a known entity—so for any new kinase inhibitor it is a fair question to ask what its selectivity is. Does it inhibit just Jaks or other kinases as well? How specific is it among the Jaks? These are important questions for any new drug coming along in order to understand its mechanism of action but also its side effects. The present Jakinibs all block more than one Jak, so all inhibit multiple cytokines. The question going forward is whether more selective Jakinibs will be as effective and potentially safer. While one might assume that more selectivity would be better, this assumption is not always borne out. Just look at the experience with non-steroidal anti-inflammatory drugs and selective Cox2 inhibitors. Another possible scenario is that multikinase inhibitors might be useful in early phases of disease treatment, when a plethora of inflammatory responses are raging. Later, when disease is more controlled, perhaps a more selective inhibitor might be safe and effective for maintenance therapy.
Lessons learned?
Thanks to the completion of the human genome, there are hundreds of potential therapeutic targets for autoimmune disease. And yet, the cost of generating a new drug typically runs to a billion or so dollars. The development of Jakinibs will surely be studied to see if there are lessons that might be gleaned for other classes of new drugs. In contrast to initial views, kinases turn out to be very ‘druggable’ and genetic information unequivocally established the requisite function of Jaks in cytokine signalling. However, knocking out Jak2 in mice resulted in embryonic lethality, so one might have thought that a drug with Jak2 activity would be problematic. It is clear that equating drugs to knockouts is not always useful. If Jaks are good targets, one might imagine that STATs would also be good targets; however, targeting the latter has proven to be extremely difficult.
Conclusions
The development of kinase inhibitors has offered new therapies for diverse clinical entities ranging from malignancy to autoimmunity. Jak inhibitors or Jakinibs initially launched to treat a rare haematological disorder are now progressing to be used in not only malignancies but common autoimmune disorders as well. The role of Jak inhibitors in the treatment algorithm of diseases ranging from the vasculitides to systemic lupus erythematosus or polymyalgia rheumatica remains to be determined. Where Jakinibs will fit in the spectrum of therapeutic options from DMARDs and steroids to biologicals and cyclophosphamide is unknown. However, the excitement is that if approved, Jakinibs will be the first new approved oral therapy for RA in a decade.
References
Footnotes
Handling editor Tore K Kvien
-
Competing interests JJO'S and the National Institutes of Health (NIH) hold patents related to targeting JAKs as targets for immunomodulatory agents. JJO'S and the NIH have a Collaborative Research Agreement and Development Award with Pfizer.
-
Provenance and peer review Not commissioned; externally peer reviewed.