
Abstract
Multicentric osteolysis, nodulosis and arthropathy (MONA) is a rare autosomal recessive disorder. To date, 13 mutations of the matrix metalloproteinase 2 (MMP2) gene have been detected in 26 patients with MONA and other osteolytic syndromes. Here, we describe the molecular and functional analysis of a novel MMP2 mutation in two adult Italian siblings with MONA. Both siblings displayed palmar-plantar subcutaneous nodules, tendon retractions, limb arthropathies, osteolysis in the toes and pigmented fibrous skin lesions. Molecular analysis identified a homozygous MMP2 missense mutation in exon 8 c.1228G>C (p.G410R), not detected in 260 controls and predicted by several bioinformatic tools to be pathogenic. By protein modelling, the mutant residue was predicted to affect the main chain conformation of the catalytic domain. Gelatin zymography, the gold standard test for MMP2 function, of serum-free conditioned medium from G410R-MMP2-expressing human embryonic kidney (HEK) cells, showed a complete loss of gelatinolytic activity. The novel mutation is located in the catalytic domain, as are 3 (p.E404K, p.V400del and p.G406D) of the other 13 MMP2 mutations described to date; however, p.G410R underlies a phenotype that is only partially overlapping that of other MMP2 exon 8 mutation carriers. Our results further delineate the complexity of genotype–phenotype correlations in MONA, broaden the repertoire of reported MMP2 mutation and enhance the comprehension of the protein motifs crucial for MMP2 catalytic activity.
Similar content being viewed by others
Introduction
Multicentric osteolysis, nodulosis and arthropathy syndrome (MONA; Online Mendelian Inheritance in Man (OMIM) #259600) is an extremely rare and progressive skeletal disorder with an autosomal recessive pattern of inheritance. It is characterised by prominent involvement of the bones, including diffuse osteoporosis, osteolysis, particularly in the hands and feet, and arthropathies with progressive multiple joint contractures. Other clinical features, such as subcutaneous fibrotic nodules, cutaneous pigmented lesions, corneal opacities, cardiac defects and facial dysmorphic features, are also variably observed.1, 2, 3, 4, 5
In 2001, Martignetti et al.6 first identified homozygous mutations in the matrix metalloproteinase 2 (MMP2) gene in two Saudi Arabian families affected by MONA. The MMP2 gene maps to chromosome 16p12.2 and encodes the 72-kDa gelatinase A protein, responsible for the recognition, binding and proteolysis of denatured type IV collagen and gelatin molecules in the extracellular matrix. MMP2 is constitutively expressed in smooth muscle cells, adipocytes, macrophages and fibroblasts. In addition to its role in extracellular matrix turnover, it is also involved in the activation of growth factors and cytokines and in cell adhesion processes, playing an important role in various complex diseases, including cancer.7,8
To date, only 13 mutations, spread across the entire coding sequence of the gene, have been identified in 26 individuals from 13 families, almost all of whom are children of consanguineous parents.4, 5, 6,9, 10, 11, 12, 13, 14, 15, 16 The mutations are private and, with only one exception, also homozygous. Individuals exhibit the onset of symptoms between the early postnatal period and ∼6 years of age and display a phenotype characterised by a certain degree of heterogeneity, because of some of the main features such as subcutaneous nodules, growth restriction, corneal alterations and cardiac malformations, not always being present. The variegated clinical presentation falls within a wide phenotypic spectrum that includes Torg syndrome, first described by Torg et al.17 in 1969. Owing to its phenotypic overlap with Torg syndrome and MONA, the clinically related Winchester syndrome (OMIM #277950), identified by Winchester et al.18 in 1969, was also once classified as an allelic osteolytic disorder.9,12,19 However, Evans et al.20 have recently distinguished it as a separate disease based on the identification, in the original Winchester patients, of a causative homozygous mutation of the type 1 transmembrane metalloproteinase MMP14 gene.
Our study focusses on two adult Italian siblings, one female and one male aged 43 and 37 years, respectively, referred to our department because of progressive osteoarticular anomalies that had led to the functional limitation of multiple joints, especially in the upper limbs. We collected detailed clinical histories and performed a clinical evaluation of both siblings. Based on their family history and observation of phenotypic features resembling those of MONA–Torg syndromes, we investigated MMP2 as a candidate causative gene and performed mutation screening of the coding sequence followed by functional analysis of the mutant MMP2 protein.
Materials and Methods
Mutation analysis
After obtaining informed consent from both siblings, genomic DNA was extracted from the patients’ whole blood samples (Promega DNA blood kit; Promega, Madison, WI, USA). Exons, intron/exon boundaries and flanking 5′ promoter and 3′ untranslated regions of the MMP2 gene (RefSeq accession numbers: NG_008989 and NM_004530) were amplified by PCR (primers and annealing temperatures are provided in Supplementary Table 1) and directly sequenced in both directions using the ABI BigDye Terminator Sequencing Kit (Applied Biosystems, Foster City, CA, USA) and an ABI3100 Genetic Analyser (Applied Biosystems). Control DNA samples from healthy individuals were screened by PCR using a pair of primers including a mutant allele-specific oligonucleotide.
In silico predictions
Evolutionary conservation of the mutation site and the affected protein residue, as well as its pathogenic potential, were evaluated using different bioinformatic prediction tools. ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/) allows the alignment of multiple sequences in different species; PolyPhen2 (http://genetics.bwh.harvard.edu/pph/) is a function algorithm based on amino acid alignment and protein structural attributes; SIFT (http://sift.jcvi.org/) and Pmut (http://mmb2.pcb.ub.es:8080/PMut/) predict pathogenic amino acid changes based on amino acid sequence alignment and neural network analysis. Additional algorithms were used, in particular Mutation Taster (http://www.mutationtaster.org/), Mutation Assessor (http://mutationassessor.org/) and Panther (http://www.pantherdb.org/; version 8.1, June 2013), a collection of protein families and subfamilies that allows the prediction of whether a given coding variant would determine a deleterious functional change. The default parameters were used for each program.
Modelling of the mutant protein
The catalytic domain of the mutant protein (residues 110–218 and 393–450) was modelled with MODELLER 9.13,21 using as a template the structure 3ayu.22 This template contains a peptide ligand that is the β-amyloid precursor protein-derived inhibitor. Active site residues were identified using the DrosteP algorithm.23 Dihedral angles and secondary structure were assigned with SEGNO.24 The effect of mutations on protein stability was assessed using SDM.25 Molecular graphics were generated with the UCSF Chimera package.26
Cloning, mutagenesis and transfection
Full-length MMP2 complementary DNA (cDNA) from control primary fibroblasts was amplified by PCR and cloned into a pcDNA3.1 vector (Invitrogen, Paisley, UK). Mutagenesis was performed using a QuikChange Site-Directed Mutagenesis Kit according to the manufacturer’s instructions (Agilent Technologies, Santa Clara, CA, USA); the c.1228 G>C substitution was produced using a pair of mismatch-introducing primers. Human embryonic kidney 293 T cells (HEK293T), grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 100 U ml−1 penicillin–streptomycin at 37 °C in a 5% CO2 atmosphere, were used for transfection. HEK cells were seeded onto Petri dishes and grown for 1 day to achieve 80% confluency. Conditioned medium (CM) was replaced with serum-free CM and then transfected with 2 μg plasmid DNA using X-tremeGENE HP DNA Transfection Reagent (Roche Diagnostic, Manheim, Germany), according to the manufacturer’s instructions. At 24 h after transfection, CM was collected for functional analysis and cells were harvested to extract RNA for transcript analysis.
RNA extraction and reverse transcriptase-PCR
Total RNA was isolated from cell lines using TRI reagent (Sigma, St. Louis, MO, USA) and treated with RNase-free DNase I (New England Bio-Labs, Ipswich, MA, USA), according to the manufacturers’ protocols. The cDNA was synthesised from 500 ng of total RNA from transfected and nontreated cell lines using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) with random hexamers. Transcript analysis was performed by MMP2 PCR amplification of both mutant and wild-type (WT) cDNA followed by direct sequencing.
Immunoblot analysis
CM (12 μl) from nontransfected and WT- and mutant-transfected cultures was mixed with nonreducing SDS Loading Buffer (Blue Loading Buffer Pack, Cell Signaling Technology, Beverly, MA, USA) and denatured at 99 °C for 3 min. Proteins were separated on 10% SDS polyacrylamide gels and subsequently transferred by electrophoresis to a polyvinylidene fluoride membrane (Roche). The membranes were then washed twice in phosphate-buffered saline (PBS) with Tween-20 (PBS-T), and nonspecific binding was blocked by incubating the membranes in 5% skimmed milk in PBS-T for 1 h at room temperature with agitation. Membranes were incubated at 4 °C overnight with agitation in rabbit anti-MMP2 antibody (sc-10736; Santa Cruz Biotechnology, Santa Cruz, CA, USA) diluted 1:500 in PBS-T. Membranes were then washed four times in PBS-T and incubated at room temperature for 1 h with agitation in horseradish peroxidase-conjugated goat anti-rabbit IgG secondary antibody (sc-2004; Santa Cruz Biotechnology) diluted 1:10 000 in PBS-T. After four washes in PBS-T and two washes in PBS, bound antibodies were detected using enhanced chemiluminescence (Westar ηC Cyanagen, Bologna, Italy). Blot images were acquired with a Gbox Chemi XT4 system (Syngene, Cambridge, UK). All experiments were performed in duplicate at least twice.
Gelatin zymography
Zymography was performed to analyse the enzymatic activity of MMP2. Serum-free CM was diluted 1:2 with nonreducing Tris-glycine sample buffer and electrophoresed for 90 min on a 10% gelatin zymogram gel (Ready Gel Zymogram Precast Gels; Bio-Rad Laboratories, Hercules, CA, USA) that was developed overnight as previously described.27 Gels were stained for 1 h with Coomassie Brilliant Blue G-250 (Bio-Rad) and then destained for 30 min. Images were acquired using a Gbox Chemi XT4 system (Syngene).
Results
Clinical report
Two Italian siblings, a female and a male aged 43 and 37 years, respectively, were referred to the Orthopaedics Division of our department with progressive osteoarticular problems. At a clinical follow-up, evaluation of the family history by a medical geneticist revealed that the two siblings were the eldest and youngest children of healthy consanguineous (second cousins) parents who had a healthy daughter and had experienced a history of miscarriages (Figure 1a). Both siblings presented with partially overlapping clinical symptoms that appeared in infancy and became progressively more severe with age. Both had experienced a normal perinatal period and both displayed normal psychomotor development.

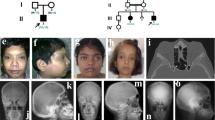
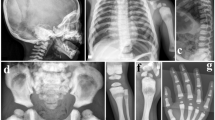
Pedigree of the study family, main clinical features of the affected siblings and radiographic findings of patient II-1. (a) Pedigree highlighting parental consanguinity (second cousins) and several miscarriages. (b–e) Primary clinical features of II-1 (b, c, d, e) and II-3 (b′, c′, d′): white arrows indicate the palmar-plantar subcutaneous nodules (b, b′, c), accompanied by swelling of the digits and prominent flexion contractures of all fingers but not the thumbs (b, b′); the feet of both patients appear shortened with deformities of the toes and cutaneous achromic plaques on the dorsal surface (c, c′); fibrous pigmented lesions can be observed on the surface of the limbs (d, d′); relapsing corneal pterygium is displayed by II-1 (e). (f) X-rays from patient II-1 at different ages; (f, top panel) both hands at age 8, 15 and 36 years show the progressive flexion of the fingers and the altered structure of the bones, with cortical thinning and trabecular texture reduction; (f, bottom panel) spinal X-rays showing dorsal scoliosis, arthrosic alterations and diffuse osteopenia; right hip at 32 and 42 years of age, showing a progressive narrowing of the articular space as seen in osteoarthritis; right forefoot at 8 years, where a fracture of the third metatarsal bone with abundant callus can be seen; the same foot at 42 years with evident osteoporotic alterations of the trabecular bone, degenerative changes in the small joints and osteolysis of part of the first and second phalanx in the fourth and fifth toes. A full colour version of this figure is available at the Journal of Human Genetics journal online.
In the affected female (II-1), cutaneous dyschromic-fibrotic striae, localised first at the trunk and then at the surface of the limbs, became apparent during infancy, followed by the development of a subcutaneous nodule on the plantar surface of the big toe at the age of 5 to 6 years. Upper limb osteopenia was revealed by X-rays following a metacarpal fracture caused by minor injury. After the age of 18 years, additional subcutaneous nodules appeared on the palmar surface of the hands, along with progressive bilateral tendon retractions. During adulthood, she developed dorsal scoliosis and suffered from progressive diffuse joint pain. After the age of 35 years, she underwent three rounds of eye surgery to treat bilateral relapsing pterygium. At the last cardiac evaluation, a first-degree atrioventricular block without evident structural defects was observed.
In the affected male (II-3), radiography had revealed osteoporosis of both the upper and lower limbs by the age of 3 years. When aged 6 years, he developed subcutaneous nodules on the palmar-plantar areas and the extensor surfaces of the elbows. After the age of 8 years, he suffered from progressive tendon retractions, localised mainly to the hands, that were treated surgically without benefit. During adolescence, he experienced two femoral fractures, the second one subsequent to minor trauma, and when aged 20 years, he developed cutaneous fibrotic plaques and striae along with progressive generalised joint stiffness. Cardiovascular evaluations performed since the age of 15 years revealed early-onset arterial hypertension.
At the last evaluation, the main clinical signs displayed by the two siblings included painful subcutaneous fibrous nodules on the hands and feet (Figure 1b, b′ and c), cutaneous achromic plaques (Figure 1c and c′), hyperpigmented fibrotic striae (Figure 1d and d′), prominent tendon retractions on both hands, functional limitation of all upper limb joints and bone resorption of the toes. Radiographic findings in patient II-1, at different ages, show progressive flexion of fingers and altered structure of the hand bones, with cortical thinning and trabecular texture reduction (Figure 1f, top panel). Other radiographic images of patient II-1 demonstrate dorsal scoliosis, evident osteoporotic alterations of the trabecular bones of the feet, with degenerative changes in the small joints and osteolysis of multiple phalanges (Figure 1f, bottom panel). Histological examination of a biopsy specimen from a subcutaneous nodule showed fibroblast proliferation. The facial appearance of both siblings was slightly coarsened (not shown), but no particular dysmorphic features or any significant stature impairment were noticed. The affected female displayed corneal pterygium (Figure 1e) that was not present in her brother.
Molecular characterisation
Based on the clinical phenotype shared by the siblings, the MMP2 gene, mapping at 16q12.2, was identified as a plausible candidate causative gene. Consistent with the hypothesis that the 16q12.2 chromosomal region in the affected siblings would host an identical mutant allele inherited by their consanguineous parents from a common ancestor, analysis of the short tandem repeats flanking MMP2 revealed that the siblings shared a homozygous genotypic pattern (Supplementary Figures 1a and b). Based on this finding, mutation screening was performed by direct sequencing of all MMP2 exons, resulting in the identification of a novel homozygous missense mutation in exon 8: c.1228G>C (p.G410R) (Supplementary Figure 1c). The mutation is not present in the Exome Variant Server (http://evs.gs.washington.edu/EVS/) or dbSNP (http://www.ncbi.nlm.nih.gov/SNP/) databases. Amplification of MMP2 exon 8 using mutation-specific primers excluded the presence of this nucleotide change in both alleles from 260 healthy controls.
In silico predictions
PolyPhen2 predicted that the identified MMP2 mutation might be damaging with the maximum score of 1. The other algorithms Mutation Taster, SIFT and Pmut provided a fully concordant prediction of the potential of this mutation to result in disease: SIFT predicted that p.G410R would not be tolerated, and Pmut ranked it as pathological. Panther provided a substitution position-specific evolutionary conservation score of −8.74 (range from 0 to −10), which is associated with a likely deleterious effect.
Modelling of the mutant MMP2 protein
The amino acid 410 of MMP2 and its flanking residues are highly evolutionarily conserved as shown by ClustalW2 alignment (Figure 2a). Although p.G410R is classified as pathogenic by all the prediction programs, they do not explain why the mutation is damaging. For this reason, we built a model of the MMP2 catalytic domain in the presence of a ligand peptide. We define the active site residues as those found in the most conserved pocket.23 These we identified as: G189–H193, L399–V400, H403–E404, H413, P417–T426, T428 and F431–S434. G410 does not belong to this group; nonetheless, it plays a pivotal role in maintaining the geometry of the active site. The model showed that G410 is in a tight turn between the long α-helix 397–409 and the short strand 411–412 (Figure 2b). The ligand peptide lies on the helix and the catalytic Zn2+ is bound by H403, H407 (within the helix) and H413 at the tip of the strand. G410 has a conformation (positive ϕ angle) that is forbidden to other residues such as arginine. Hence, the mutation affects the main chain conformation of the turn and, as a consequence, the active site. We cannot exclude that the mutation p.G410R affects stability too because the program SDM,25 which predicts changes in the stability of proteins caused by mutations, assigns a negative score to G410R.

Phylogenetic conservation and position within the matrix metalloproteinase 2 (MMP2) catalytic domain of the affected G410 residue. (a) ClustalW multiple sequence alignment showing a high degree of conservation of wild-type Gly410 and flanking residues among different species. (b) The structure of the p.G410R catalytic domain obtained by homology modelling: it is shown as cartoon, along with the side chains of residue R410 and residues that bind the catalytic Zn2+; dark blue is used to highlight the α-helix 397–409 and the strand 411–412; the peptide ligand is shown in green.
Functional analysis
To demonstrate the effect of the c.1228G>C missense mutation, present on both alleles, we performed serum gelatin zymography, a standard assay for MMP2 catalytic activity. However, additional patient blood samples were not available. Therefore, we cloned WT MMP2 cDNA from control fibroblasts and applied site-directed mutagenesis to generate the c.1228G>C allele. We then transfected WT and mutant cDNA into recipient HEK cells and assayed the CM for gelatinolytic function. Transcript analysis confirmed the overexpression of each transfected cDNA (data not shown). Western blot analysis confirmed the presence of MMP2 protein in the CM collected from both WT and mutant cDNA-transfected HEK cells, whereas any endogenous MMP2 in the CM of untreated HEK cells was below detectable levels (Figure 3a). To assess gelatinolytic activity, samples of serum-free CM were analysed by gelatin zymography. As shown in Figure 3b, CM from control fibroblasts, control serum and WT MMP2-transfected HEK cells showed a clear gelatinolytic band, whereas CM from c.1228C-MMP2-transfected HEK cells demonstrated a complete loss of enzymatic activity.

Western blot and zymography of wild-type (WT) and G410R-mutant matrix metalloproteinase 2 (MMP2) protein. (a) Western blot of conditioned media (CM) from human embryonic kidney (HEK) cells with an anti-MMP2 antibody shows the 72-kDa band in cells transfected with the WT vector or the mutant (MUT) G410R-vector; no band can be seen in nontreated (NT) HEK cells. (b) The proteins to be assessed with zymography are separated by electrophoresis in a polyacrylamide gel containing a specific substrate (gelatin) that is co-polymerised with the acrylamide under nonreducing and denaturing conditions. After renaturation and incubation with the activation buffer, the concentrated MMPs in the gel are able to digest the substrate. The gel is then stained with Coomassie blue and the active MMPs are detected as clear bands against a dark background of undegraded substrate. The gelatinolytic assay shows activity in control serum, fibroblast (FIB) and WT-HEK CM; no activity is detected in CM from NT HEK cells and HEK cells transfected with the MUT vector.
Discussion
The MMP2 mutations that have been identified so far are spread throughout the gene and affect all the main protein domains. Out of 14 mutations, 9 are clustered within the catalytic domain of the protein, and half of them, including c.1228G>C, are located in exon 8. Missense mutations are the most frequently observed type (seven), followed by truncating mutations (three nonsense and two frameshift), one splicing mutation (c.658+2T>C) that leads to the skipping of exon 4 and one small in-frame deletion (c.1488_1490delTGG, p.V400del).
All mutations appear to be private and homozygous, with the exception of p.R101H that affects the residue adjacent to Cys102 responsible for the MMP2 ‘cysteine-switch’ activation mechanism.28,29 This mutation was first identified in a Saudi Arabian family by Martignetti et al.6 in 2001, and then in a girl from the United States,10 who is the only compound heterozygous mutation carrier reported to date. Interestingly, the same residue was recently found to be altered by a different mutation (p.R101C) in a Moroccan boy,14 indicating that Arg101 is crucial for the proper functional activation of the MMP2 protein. All of the reported mutations appear to interfere with MMP2 catalytic function, leading to the loss of gelatinolytic activity, as has been demonstrated by gelatin zymographic analysis of half (seven) of the mutations.5,6,10,12,14 With respect to the p.G410R mutation described here, we have ruled out its presence in 260 healthy controls and have interrogated several bioinformatics algorithms, all of which predicted that this mutation is damaging. The in vitro overexpression of both WT and G410R-MMP2 allowed us to not only demonstrate that the mutant MMP2 does not retain its main biological activity (Figure 3b) but also to confirm that the altered protein is synthesised, as shown by immunoblot analysis of the CM from mutant MMP2-transfected HEK cells (Figure 3a), in agreement with the detection of the overexpressed mutant transcript. The obtained evidence is clearcut, because HEK cells produce barely detectable amounts of endogenous MMP2 protein compared with both WT- and mutant-transfected HEK cell lines (Figure 3a). Moreover, analysis and comparison of the CM of HEK cells indicates that the G410R mutant protein is not only synthesised but also exported. Although it remains unknown whether there is a quantitative difference between the amount of G410R-MMP2 produced and the amount exported, the apparent reduction, seen by western blotting, of the overexpressed mutant protein in CM, compared with the overexpressed WT MMP2, could be an indicator of altered stability of the mutant protein, as predicted by the SDM software.
Assay of gelatinolytic activity, the gold standard test for MMP2 function, demonstrated that the novel missense mutation observed in the siblings caused loss of function. This finding underlines the pathogenic role of the mutation, allowing us to reexamine the patients’ phenotypic presentation for peculiarities and similarities to other reported patients. Rather than being characterised at a young age, like most MONA cases reported in the literature, the siblings described herein were evaluated after more than 30 years with the disease; however, as for all reported patients, the onset of the syndrome occurred at preschool age.
Both siblings displayed the main hallmarks of the syndrome: progressive distal arthropathy, which led to functional impairment of multiple joints, flexion contractures, which were markedly debilitating in the upper limbs, and diffuse osteopenia with osteolysis, especially evident in the tarsal and phalangeal bones. In addition, they displayed other common signs, although not always observed, including painful subcutaneous nodules in association with a reported worsening of the flexion contractures of the hands and feet and hyperpigmented fibrotic hardening of the cutis that appeared during infancy in II-1 and after adolescence in II-3.
Although the siblings show a largely overlapping phenotype, intrafamilial variability is attested by some of the clinical features of II-1 that are not present in II-3, including dorsal scoliosis, electrocardiographic alterations and ophthalmologic involvement. Differences in the clinical expressivity of the same mutant gene observed in these patients suggest that the phenotype might be modulated by modifier genes, as observed in other Mendelian diseases.30
To the best of our knowledge, the parental recurrent miscarriages reported by our patients have not been described in other MONA, Torg or Winchester syndrome families. Although this finding should be considered with caution, as other uninvestigated genetic causes cannot be ruled out, numerous reports have shown that MMPs play a role in a variety of both physiological and pathological pregnancy-associated processes, and it has been hypothesised that increased levels of circulating gelatinases are associated with spontaneous early pregnancy failure.31
Correlations between genotype and phenotype in MONA are difficult to determine because all functionally tested mutations apparently abolish MMP2 proteolytic activity, thereby making it hard to link the underlying molecular mechanism to the highly variable clinical features. Jeong et al.12 observed that bearers of homozygous mutations within the MMP2 catalytic domain may display a distinct phenotype characterised by the absence of subcutaneous nodules and wide metacarpals. The mutation identified in the present study is localised to the same peptide region as three other pathogenic mutations (p.V400del, p.E404K and p.G406D) within a highly conserved region of the catalytic domain that plays a critical role in the peptidase activity, being the long α-helix 397–409 where the ligand lies (Figure 2b). However, unlike p.V400del, p.E404K and p.G406D, p.G410R is associated with multiple prominent subcutaneous nodules and the absence of evident growth restriction (Table 1). Conversely, according to both the clinical description of the reported cases, recently reviewed by Castberg et al.,14 and the phenotypic features of our patients, no congenital heart malformation has been found to be associated with any missense mutation or small in-frame deletion localised within the catalytic domain. The identification of additional MMP2 mutations in MONA-affected individuals could allow to infer the impact on the clinical phenotype resulting from mutations that affect the same protein domain.
In conclusion, with the addition of two sibling patients, our study enlarges the cohort with the extremely rare MONA syndrome who can be followed up as the disease evolves and thus will help to clarify the clinical definition. The clinical evaluation of the two siblings highlights the complexity of genotype–phenotype correlations in MONA, as well as the variability in intrafamilial expressivity, that could be ascribed to other genetic or environmental factors. Furthermore, our report widens the repertoire of reported MMP2 mutation and documents the loss of the main biological function of the G410R mutant protein, highlighting residues and motifs crucial for the catalytic activity.
References
Eisenstein D. M ., Poznanski A. K . & Pachman L. M . Torg osteolysis syndrome. Am. J. Med. Genet. 80, 207–212 (1998).
Al-Mayouf S. M ., Majeed M ., Hugosson C . & Bahabri S . New form of idiopathic osteolysis: nodulosis, arthropathy and osteolysis (NAO) syndrome. Am. J. Med. Genet. 93, 5–10 (2000).
Al Aqeel A ., Al Sewairi W ., Edress B ., Gorlin R. J ., Desnick R. J . & Martignetti J. A . Inherited multicentric osteolysis with arthritis: a variant resembling Torg syndrome in a Saudi family. Am. J. Med. Genet. 93, 11–18 (2000).
Zankl A ., Bonafé L ., Calcaterra V ., Di Rocco M . & Superti-Furga A . Winchester syndrome caused by a homozygous mutation affecting the active site of matrix metalloproteinase 2. Clin. Genet. 67, 261–266 (2005).
Tuysuz B ., Mosig R ., Altun G ., Sancak S ., Glucksman M. J . & Martignetti J. A . A novel matrix metalloproteinase 2 (MMP2) terminal hemopexin domain mutation in a family with multicentric osteolysis with nodulosis and arthritis with cardiac defects. Eur. J. Hum. Genet. 17, 565–572 (2009).
Martignetti J. A ., Aqeel A. A ., Sewairi W. A ., Boumah C. E ., Kambouris M ., Mayouf S. A . et al. Mutation of the matrix metalloproteinase 2 gene (MMP2) causes a multicentric osteolysis and arthritis syndrome. Nat. Genet. 28, 261–265 (2001).
Gupta G. P ., Nguyen D. X ., Chiang A. C ., Bos P. D ., Kim J. Y ., Nadal C . et al. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature 446, 765–770 (2007).
Cheung P. Y ., Sawicki G ., Wozniak M ., Wang W ., Radomski M. W . & Schulz R . Matrix metalloproteinase-2 contributes to ischemia-reperfusion injury in the heart. Circulation 101, 1833–1839 (2000).
Rouzier C ., Vanatka R ., Bannwarth S ., Philip N . & Coussement A ., Paquis-Flucklinger V . et al. A novel homozygous MMP2 mutation in a family with Winchester syndrome. Clin. Genet. 69, 271–276 (2006).
Zankl A ., Pachman L ., Poznanski A ., Bonafé L ., Wang F ., Shusterman Y . et al. Torg syndrome is caused by inactivating mutations in MMP2 and is allelic to NAO and Winchester syndrome. J. Bone Miner. Res. 22, 329–333 (2007).
Gok F ., Crettol L. M ., Alanay Y ., Hacihamdioglu B ., Kocaoglu M ., Bonafe L . et al. Clinical and radiographic findings in two brothers affected with a novel mutation in matrix metalloproteinase 2 gene. Eur. J. Pediatr. 169, 363–367 (2010).
Jeong S. Y ., Kim B. Y ., Kim H. J ., Yang J. A . & Kim O. H . A novel homozygous MMP2 mutation in a patient with Torg-Winchester syndrome. J. Hum. Genet. 55, 764–766 (2010).
Temtamy S. A ., Ismail S ., Aglan M. S ., Ashour A. M ., Hosny L. A ., El-Badry T. H . et al. A report of three patients with MMP2 associated hereditary osteolysis. Genet. Couns. 23, 175–184 (2012).
Castberg F. C ., Kjaergaard S ., Mosig R. A ., Lobl M ., Martignetti C ., Martignetti J. A . et al. Multicentric osteolysis with nodulosis and arthropathy (MONA) with cardiac malformation, mimicking polyarticular juvenile idiopathic arthritis: case report and literature review. Eur. J. Pediatr. 172, 1657–1663 (2013).
Ekbote A. V ., Danda S ., Zankl A ., Mandal K ., Maguire T . & Ungerer K . Patient with Mutation in the Matrix Metalloproteinase 2 (MMP2) Gene - A Case Report and Review of the Literature. J. Clin. Res. Pediatr. Endocrinol. 6, 40–46 (2014).
Leiden Open Variation Database (MMP2 homepage). https://grenada.lumc.nl/LOVD2/mendelian_genes/home.php?select_db=MMP2.
Torg J. S ., DiGeorge A. M ., Kirkpatrick J. A. Jr & Trujillo M. M . Hereditary multicentric osteolysis with recessive transmission: a new syndrome. J. Pediatr. 75, 243–252 (1969).
Winchester P ., Grossman H ., Lim W. N . & Danes B. S . A new acid mucopolysaccharidosis with skeletal deformities simulating rheumatoid arthritis. Am. J. Roentgenol. Radium. Ther. Nucl. Med 106, 121–128 (1969).
Warman M. L ., Cormier-Daire V ., Hall C ., Krakow D ., Lachman R ., LeMerrer M . et al. Nosology and classification of genetic skeletal disorders: 2010 revision. Am. J. Med. Genet. A 155A, 943–968 (2011).
Evans B. R ., Mosig R. A ., Lobl M ., Martignetti C. R ., Camacho C ., Grum-Tokars V . et al. Mutation of membrane type-1 metalloproteinase, MT1-MMP, causes the multicentric osteolysis and arthritis disease Winchester syndrome. Am. J. Hum. Genet. 91, 572–576 (2012).
Marti-Renom M. A ., Stuart A. C ., Fiser A ., Sanchez R ., Melo F . & Sali A . Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct. 29, 291–325 (2000).
Hashimoto H ., Takeuchi T ., Komatsu K ., Miyazaki K ., Sato M . & Higashi S . Structural basis for matrix metalloproteinase-2 (MMP-2)-selective inhibitory action of beta-amyloid precursor protein-derived inhibitor. J. Biol. Chem. 286, 33236–33243 (2011).
Cammisa M ., Correra A ., Andreotti G . & Cubellis M. V . Identification and analysis of conserved pockets on protein surfaces. BMC Bioinformatics 14 (suppl. 7), S9 (2013).
Cubellis M. V ., Cailliez F . & Lovell S. C . Secondary structure assignment that accurately reflects physical and evolutionary characteristics. BMC Bioinformatics. 6 (suppl. 4), S8 (2005).
Worth C. L ., Preissner R . & Blundell T. L . SDM—a server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 39 (Web Server issue), W215–W222 (2011).
Pettersen E. F ., Goddard T. D ., Huang C. C ., Couch G. S . & Greenblatt D. M ., Meng E. C . et al. UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Hawkes S. P ., Li H . & Taniguchi G. T . Zymography and reverse zymography for detecting MMPs and TIMPs. Methods Mol. Biol. 622, 257–269 (2010).
Morgunova E ., Tuuttila A ., Bergmann U ., Isupov M ., Lindqvist Y ., Schneider G . et al. Structure of human pro-matrix metalloproteinase-2: activation mechanism revealed. Science 284, 1667–1670 (1999).
Hadler-Olsen E ., Fadnes B ., Sylte I ., Uhlin-Hansen L . & Winberg J. O . Regulation of matrix metalloproteinase activity in health and disease. FEBS J. 278, 28–45 (2011).
Nadeau JH . Modifier genes in mice and humans. Nat. Rev. Genet. 2, 165–174 (2001).
Nissi R ., Talvensaari-Mattila A ., Kotila V ., Niinimäki M ., Järvelä I . & Turpeenniemi-Hujanen T . Circulating matrix metalloproteinase MMP-9 and MMP-2/TIMP-2 complex are associated with spontaneous early pregnancy failure. Reprod. Biol. Endocrinol. 11, 2 (2013).
Acknowledgements
This study was supported by ‘Accordo quadro Università-Regione Lombardia n. 17292’ to L.L. and by PUR09 (Programma dell’Università per la Ricerca–10% dote giovani ricercatori) to C.G. (Università di Milano). We thank the patients for participating in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Journal of Human Genetics website
Rights and permissions
About this article

Cite this article
Azzollini, J., Rovina, D., Gervasini, C. et al. Functional characterisation of a novel mutation affecting the catalytic domain of MMP2 in siblings with multicentric osteolysis, nodulosis and arthropathy. J Hum Genet 59, 631–637 (2014). https://doi.org/10.1038/jhg.2014.84
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/jhg.2014.84
This article is cited by
-
Clinical, radiographic and molecular characterization of two unrelated families with multicentric osteolysis, nodulosis, and arthropathy
BMC Musculoskeletal Disorders (2023)
-
Bisphosphonates in multicentric osteolysis, nodulosis and arthropathy (MONA) spectrum disorder – an alternative therapeutic approach
Scientific Reports (2016)

{kind=link}