Article Text

Abstract
Objective A genome-wide association study (GWAS) of gout and its subtypes was performed to identify novel gout loci, including those that are subtype-specific.
Methods Putative causal association signals from a GWAS of 945 clinically defined gout cases and 1213 controls from Japanese males were replicated with 1396 cases and 1268 controls using a custom chip of 1961 single nucleotide polymorphisms (SNPs). We also first conducted GWASs of gout subtypes. Replication with Caucasian and New Zealand Polynesian samples was done to further validate the loci identified in this study.
Results In addition to the five loci we reported previously, further susceptibility loci were identified at a genome-wide significance level (p<5.0×10−8): urate transporter genes (SLC22A12 and SLC17A1) and HIST1H2BF-HIST1H4E for all gout cases, and NIPAL1 and FAM35A for the renal underexcretion gout subtype. While NIPAL1 encodes a magnesium transporter, functional analysis did not detect urate transport via NIPAL1, suggesting an indirect association with urate handling. Localisation analysis in the human kidney revealed expression of NIPAL1 and FAM35A mainly in the distal tubules, which suggests the involvement of the distal nephron in urate handling in humans. Clinically ascertained male patients with gout and controls of Caucasian and Polynesian ancestries were also genotyped, and FAM35A was associated with gout in all cases. A meta-analysis of the three populations revealed FAM35A to be associated with gout at a genome-wide level of significance (pmeta=3.58×10−8).
Conclusions Our findings including novel gout risk loci provide further understanding of the molecular pathogenesis of gout and lead to a novel concept for the therapeutic target of gout/hyperuricaemia.
- Gout
- Gene Polymorphism
- Arthritis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Gout is a common disease characterised by acute painful arthritis, and its global burden continues to rise with the increasingly ageing population.1 Gout is caused by hyperuricaemia, and can be classified according to patients' clinical parameters reflecting its causes2 ,3 as renal overload (ROL) gout and renal underexcretion (RUE) gout. As shown in online supplementary figure S1, patients with gout with increased urinary excretion of urate due to overproduction and/or decreased extra-renal underexcretion of urate are classified as having ROL gout, whereas those with decreased renal excretion of urate are defined as having RUE gout.2 Reflecting their causes, almost all patients with gout are divided into those two subtypes. Although these subtypes are important from both genetic and pathophysiological points of view,2 ,4 genome-wide association studies (GWASs) of gout subtypes have never been performed, partly due to the difficulty in assembling sufficient gout cases with requisite clinical data, including data from a time-consuming urinary collection examination.
supplementary data
We and other groups5–9 recently reported gout/hyperuricaemia to have relatively strong genetic risk factors. More recently, and for the first time, we performed a GWAS with only clinically defined Japanese male gout cases in which 16 single nucleotide polymorphisms (SNPs) were replicated, and five gout-risk loci were identified including two novel loci (MYL2-CUX2 and CNIH-2).10 In the present study (see online supplementary figure S2), we extended our analysis to identify novel susceptibility loci for gout by replicating approximately 2000 SNPs top-ranked in the GWASs of all gout and/or its subtypes. In addition, for the first time, we performed GWASs of gout subtypes to identify subtype-specific (cause-specific) risk loci. Furthermore, we conducted a replication study with independent Caucasian and Polynesian populations to validate loci.
Methods
Subjects and genotyping
Genome-wide genotyping was performed with the Illumina HumanOmniExpress-12 v1.0 (Illumina) platform using 946 clinically defined gout cases and 1213 controls, all Japanese males. Detailed methods of genotyping and quality control are previously described.10 Ultimately, 570 442 SNPs passed filters for 945 cases and 1213 controls. At the replication stage, 1246 cases were genotyped with a custom genotype platform using iSelect HD Custom Genotyping BeadChips (Illumina) on 1961 SNPs, as described in online supplementary methods and supplementary figure S3, and 150 gout cases were genotyped with the Illumina HumanOmniExpress-24 v1.0 (Illumina) platform. For controls, 1268 Japanese males with a serum uric acid (SUA) level ≤ 7.0 mg/dL and without gout history were recruited from BioBank Japan11 ,12 and genotyped with the Illumina HumanOmniExpress-12 v1.0 (Illumina) platform. Finally, 1961 SNPs with 1396 gout cases and 1268 controls were successfully genotyped (see online supplementary table S1). A genome-wide significance threshold was set to be α=5.0×10−8 to claim evidence of a significant association.
GWASs of the two subtypes of gout, ROL gout and RUE gout (see online supplementary figure S1), were also performed, followed by replication studies with a custom SNP chip (see online supplementary figure S3) and a subsequent meta-analysis. As described previously,2 ,10 and shown in online supplementary figure S1 and supplementary methods, ROL gout and RUE gout are defined when patients' urinary urate excretion is over 25.0 mg/hour/1.73 m2 (600 mg/day/1.73 m2) and patients' urate clearance (urate clearance/creatinine clearance ratio, FEUA) is under 5.5%, respectively. For GWASs of gout subtypes, 1178 cases were classified as ROL gout (560 cases at GWAS stage and 618 cases at replication stage) and 1315 cases as RUE gout (619 cases at GWAS stage and 696 cases at replication stage), respectively (see online supplementary table S2).
A replication study with independent Caucasian and New Zealand (NZ) Polynesian sample sets was also performed to validate the genetic risk loci identified in the present study. This replication was done in a data set recruited from New Zealand13 and from Europe by the Eurogout Consortium14 comprising 1319 male cases and 514 male controls of European ancestry and 971 male cases and 565 male controls of NZ Polynesian ancestry. SNPs were genotyped by an allelic discrimination assay (TaqMan) with a LightCycler 480 Real-Time PCR (RT-PCR) System (Roche Applied Science, Indianapolis, Indiana, USA). Detailed information of clinical characteristics and genetic analysis is shown in online supplementary methods and tables S1–S3.
Statistical analyses
The inverse-variance fixed-effects model was used for meta-analysis. In the meta-analysis with Japanese, Caucasian and NZ Polynesian populations or in the presence of heterogeneity (phet < 0.05 or I2 > 50%), we implemented the DerSimonian and Laird random-effects model for meta-analysis.15 For the replication analysis with Caucasian and NZ Polynesian sample sets, ORs were adjusted by age and ancestral group. All the meta-analyses were performed using the R V.3.1.1 and 3.2.2 (R Development Core Team. R: a language and environment for statistical computing. Vienna: R. Foundation for Statistical Computing, 2006) with meta package. All calculations of linkage disequilibrium (LD, measured in r2) were conducted using the Japanese population. The detailed information for statistical analyses is described in online supplementary methods.
Functional and localisation analyses
Urate transport analysis of NIPAL1 was performed with an oocyte expression system16 ,17 with high potassium (HK) buffer or HK buffer without magnesium. For immunohistochemical analysis, the human kidney sections (3 μm) incubated with antihuman NIPAL1 antibody (1:50) (LS-C164878; LifeSpan BioSciences, Washington, USA) or with anti-human FAM35A antibody (1:75) (HPA036582; Sigma-Aldrich, Missouri, USA) were used, and then visualised with diaminobenzidine (0.8 mM).18 ,19 Intracellular localisation of NIPAL1 was also studied in Xenopus oocytes and Madin-Darby canine kidney II (MDCKII) cells. Detailed information for the functional and localisation analyses is described in online supplementary methods.
Results
GWAS of all gout and its subtypes
In addition to the GWAS stage previously performed with 945 patients with clinically defined gout and 1213 controls, all Japanese males10 (see online supplementary figure S4), the replication stage for all cases of gout was carried out by genotyping 1961 SNPs (see online supplementary figure S3 and supplementary note) in a further 1396 male patients and 1268 male controls, and a meta-analysis then conducted (see online supplementary figure S2). Furthermore, GWASs of two subtypes of gout, ROL gout (figure 1A) and RUE gout (figure 1B), were also performed in the present study, followed by replication studies with a custom SNP chip and a subsequent meta-analysis.

Manhattan plots of GWASs of subtypes of gout. Manhattan plots of GWASs of (A) ROL gout subtype and (B) RUE gout subtype. X-axis shows chromosomal positions. Y-axis shows −log10 p values. The upper and lower dotted lines indicate the genome-wide significance threshold (p=5.0×10−8) and the cut-off level for selecting single nucleotide polymorphisms for replication study (p=0.001), respectively. GWAS, genome-wide association study; ROL, renal overload; RUE, renal underexcretion.
Meta-analysis of both the GWAS and the replication study for all gout cases (table 1) identified eight loci which showed evidence for associations at the genome-wide significance level: rs3114020 of ABCG2 (pmeta=8.66×10−35; OR=1.89), rs1014290 of SLC2A9 (pmeta=6.50×10−26; OR=1.57), rs4766566 of CUX2 (pmeta=4.03×10−20; OR=1.51), rs2285340 of SLC22A12 (pmeta=4.61×10−11; OR=1.40), rs1260326 of GCKR (pmeta=7.19×10−11; OR=1.31), rs1165176 of SLC17A1 (pmeta=1.47×10−9; OR=1.42), rs11758351 of HIST1H2BF-HIST1H4E (pmeta=1.63×10−8; OR=1.40) and rs4073582 of CNIH-2 (pmeta=3.56×10−8; OR=1.58). Among these eight loci, SLC22A12, SLC17A1 and HIST1H2BF-HIST1H4E (figure 2A–C) were first identified as gout-risk loci by the GWAS approach at the genome-wide significance level. SLC17A1 was identified here by the GWAS approach for the first time, while Hollis-Moffatt et al20 reported that rs1183201, another SNP of SLC17A1, is strongly associated with gout in Caucasians and NZ Polynesian sample sets by the candidate gene approach. While rs11758351 of HIST1H2BF-HIST1H4E is located 374 kb downstream from rs1165176 of SLC17A1, they are not in LD with each other (r2=0.03), demonstrating them to be independent susceptibility loci for gout. There was also a significant signal from rs2532941 of VARS2 (pmeta=2.74×10−8; OR=1.32), which is located downstream of HIST1H2BF-HIST1H4E by 4.7 Mb, and is reported to be associated with mitochondrial respiration.21 Since rs2532941 of VARS2 showed mild LD with rs11758351 of HIST1H2BF-HIST1H4E (r2=0.37), its significance did not remain for the GWAS stage samples after adjustment with rs11758351 of HIST1H2BF-HIST1H4E (p=0.08), or with both rs1165176 of SLC17A1 and rs11758351 (p=0.11).
Single nucleotide polymorphisms (SNPs) associated with gout and its subtypes at a genome-wide level of significance in the Japanese population

Regional association plots of five discovered loci. Three loci were revealed to exceed the genome-wide significance level from the meta-analysis with all gout cases, and two loci with renal underexcretion (RUE) gout cases. The highest association signal in each panel is located on (A) SLC22A12, (B) SLC17A1 and (C) HIST1H2BF-HIST1H4E for all gout cases, and (D) NIPAL1 and (E) FAM35A for RUE gout cases. The region within 250 kb from the single nucleotide polymorphism (SNP) indicating the lowest p value is shown. (Top panel) Plots of −log10 p values for the test of SNP association with gout in the genome-wide association study stage. The SNP showing the lowest p value in the meta-analysis is depicted as a pink diamond. Other SNPs are colour-coded according to the extent of linkage disequilibrium (measured in r2) with the SNP showing the lowest p value. (Middle panel) Recombination rates (centimorgans per Mb) estimated from HapMap Phase II data are plotted. (Bottom panel) RefSeq genes. Genomic coordinates are based on NCBI human genome reference sequence build 37.
For GWASs of gout subtypes, 1178 cases were classified as ROL gout (560 cases at GWAS stage and 618 cases at replication stage) and 1315 cases as RUE gout (619 cases at GWAS stage and 696 cases at replication stage), respectively (see online supplementary table S2). The meta-analysis of a GWAS of the ROL gout subtype and a replication study revealed significant SNPs in the following four loci: rs2728104 of ABCG2 (pmeta=5.08×10−33; OR=1.84), rs4766566 of CUX2 (pmeta=8.14×10−17; OR=1.59), rs3733589 of SLC2A9 (pmeta=2.25×10−13; OR=1.47) and rs1260326 of GCKR (pmeta=5.39×10−9; OR=1.35).
Another subtype analysis, that is, the meta-analysis of a GWAS of RUE gout and a replication study (table 1) demonstrated significant SNPs in the following seven loci: rs1014290 of SLC2A9 (pmeta=8.71×10−25; OR=1.69), rs1871744 of ABCG2 (pmeta=2.49×10−22; OR=1.81), rs4766566 of CUX2 (pmeta=2.17×10−18; OR=1.60), rs2285340 of SLC22A12 (pmeta=8.79×10−10; OR=1.44), rs780094 of GCKR (pmeta=1.62×10−9; OR=1.35), rs11733284 of NIPAL1 (pmeta=1.13×10−8; OR=1.34) and rs7903456 of FAM35A (pmeta=4.29×10−8; OR=1.34). The latter two loci, NIPAL1 and FAM35A, were novel risk loci by the GWAS of the RUE gout subtype (figure 2D, E). In total, 10 loci were identified from the present GWAS of gout and its subtypes (table 1 and see online supplementary table S4).
Of the seven loci newly identified by GWAS of the RUE gout subtype, only NIPAL1 and FAM35A had not been implicated previously in the GWASs of SUA levels or gout. Analysis with data from previously reported GWAS22 of SUA in Caucasians revealed the association with NIPAL1 and FAM35A loci (see online supplementary figure S5).
Urate transport analysis of NIPAL1 transporter
NIPAL1 and FAM35A were revealed to be associated with RUE gout in the present study. NIPAL1 has been reported to be a magnesium transporter,23 which has nine transmembrane domains (figure 3A), whereas FAM35A is predicted to be a soluble protein. In this context, we hypothesised that NIPAL1 could be involved in the regulation of urate handling as a renal urate efflux transporter. However, our functional analysis using Xenopus oocytes did not show urate transport via NIPAL1, regardless of the presence of magnesium (figure 3B).
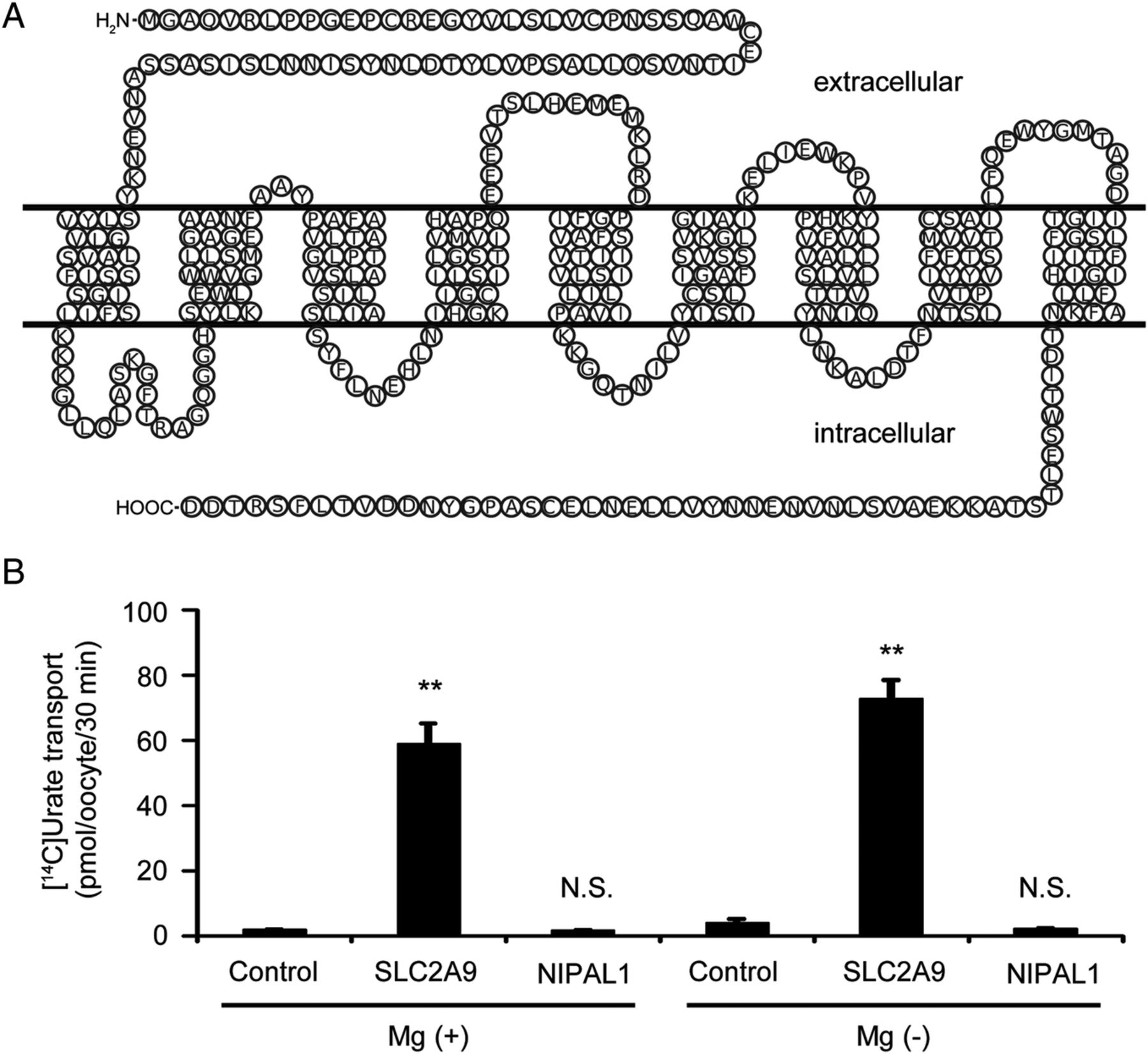
Functional analysis of NIPAL1 transporter. (A) The topological model of the NIPAL1 transporter. NIPAL1 is predicted to have nine transmembrane regions. The amino acid sequences of NIPAL1 were obtained from GenBank (accession code NM_207330). (B) Urate transport analysis of NIPAL1. SLC2A9 (also known as GLUT9) is a renal urate transporter and is used for a positive control for the urate transport analysis. In contrast to SLC2A9, urate transport via NIPAL1 was not detected, regardless of the presence of magnesium. Data are expressed as mean±SEM (n=8). Statistical analyses for significant differences were performed according to Student's t-test. (**p<0.01; N.S., not significantly different as compared with control.).
Localisation analysis of NIPAL1 and FAM35A
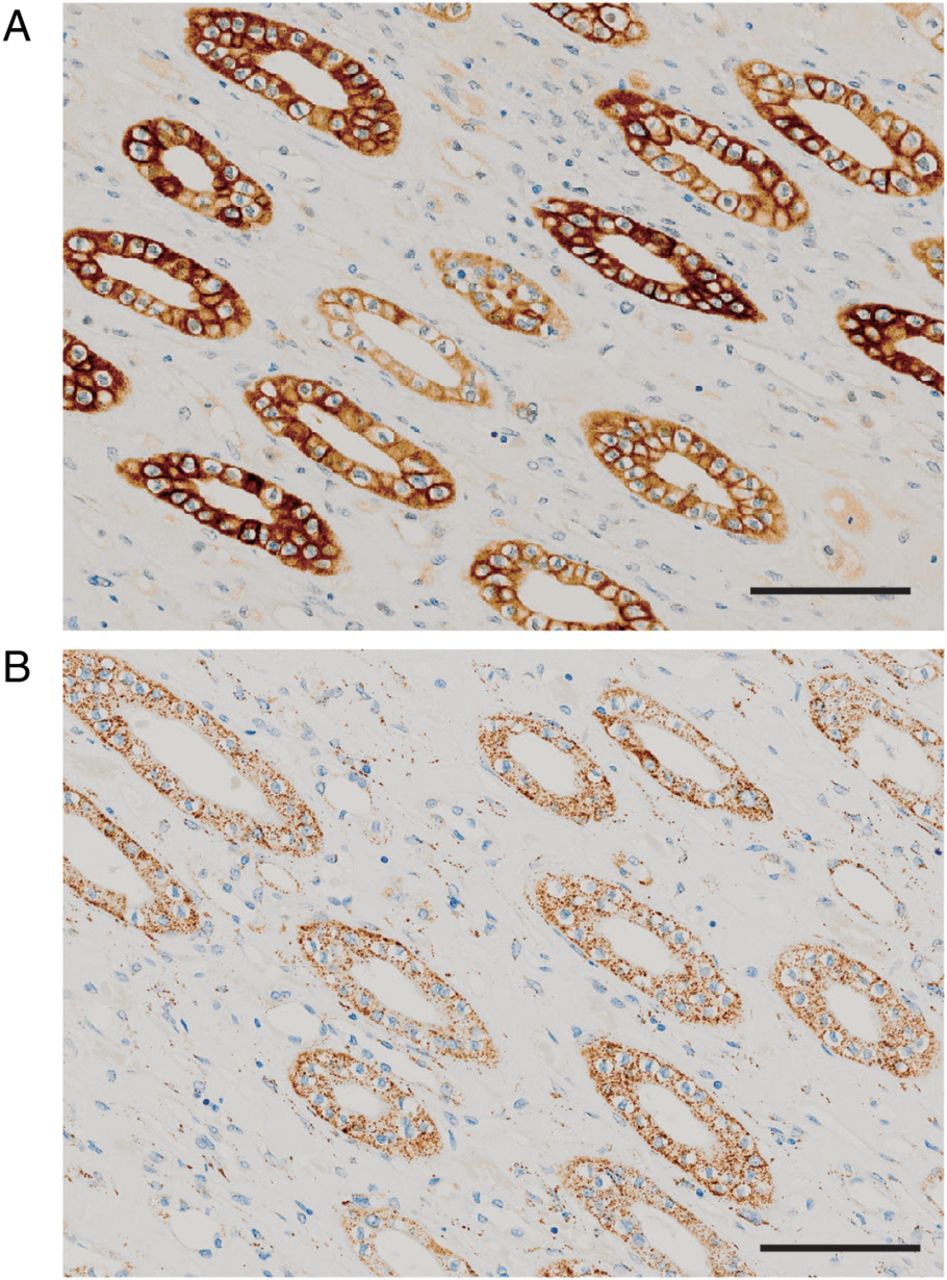
By immunohistochemical analysis, NIPAL1 and FAM35A showed cytosolic expression in the renal distal tubules of human kidney (figure 4A, B). Both proteins were also weakly detected in the cytoplasm of collecting ducts. NIPAL1-expressing Xenopus oocytes and MDCKII cells also showed intracellular localisation of NIPAL1 (see online supplementary figure S6).

Localisation analysis of NIPAL1 and FAM35A in the human kidney. Cytosolic expression was detected strongly in distal tubules and weakly in collecting ducts in human kidney for (A) NIPAL1 protein and (B) FAM35A protein. Bar=100 μm.
Replication study of all gout cases with Caucasian and Polynesian populations
A replication study for the discovered loci (SLC22A12, SLC17A1, HIST1H2BF-HIST1H4E, NIPAL1 and FAM35A) was performed for all gout cases with males drawn from Caucasian (1319 cases and 514 controls) and NZ Polynesian populations (971 cases and 565 controls). Because a gain-of-function SNP of SLC17A1, rs1165196 (Ile269Thr),16 was in strong LD with rs1165176 (r2=0.99), we performed the following analyses using rs1165196, assuming that the causal SNP in this locus was rs1165196 of SLC17A1. Among these five loci, the meta-analysis of those populations for all gout revealed a significant association with rs7903456 of FAM35A (pmeta=9.72×10−3; OR=1.17) (table 2). Although SLC17A1 did not show significance (pmeta=0.119) in the present study of those populations (table 2), a previous paper20 revealed a significant association of SLC17A1 with gout in Caucasian and NZ Polynesian sample sets, indicating the necessity of further replication studies to investigate the ancestral differences in the significance of other genetic loci including SLC17A1. Genotyping the CUX2 and CNIH-2 loci, which were identified in both our present and previous GWASs of Japanese,10 was also performed, and the CUX2 locus was replicated successfully for the first time in other populations (see online supplementary table S5). The results of further association analyses and expression quantitative trait locus (eQTL) analysis are shown in online supplementary note and tables S6 and S7. Significant effects on FEUA were detected in NIPAL1, FAM35A and SLC22A12 loci in the Japanese population, and were also observed at SLC17A1 in NZ Polynesian population.
Replication study of all gout for five discovered loci in Caucasian and NZ Polynesian sample sets
A further meta-analysis of all gout cases with Japanese, Caucasian and NZ Polynesian populations was performed for NIPAL1 and FAM35A, which were at a genome-wide significance level in the Japanese population only for the RUE gout subtype, and not for all gout cases. rs11733284 of NIPAL1 was not associated with all gout (pmeta=0.16; OR=1.11), suggesting the presence of ancestral differences in genetic effects of this locus, or a subtype-specific effect. On the other hand, rs7903456 of FAM35A showed an association with all gout at a genome-wide level of significance (pmeta=3.58×10−8; OR=1.23) (figure 5), indicating that rs7903456 is a susceptibility locus for all gout as well as the RUE gout subtype.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Forest plots for all gout among Japanese, Caucasian and New Zealand (NZ) Polynesian populations. Although rs11733284 of NIPAL1 (A) did not show significant association with all gout, rs7903456 of FAM35A (B) revealed an association with all gout at a genome-wide significance level (pmeta=3.58×10−8; OR=1.23). GWAS, genome-wide association study.
Meta-analysis of all gout for the other three loci (SLC22A12, SLC17A1 and HIST1H2BF-HIST1H4E) was also performed with Japanese, Caucasian and NZ Polynesian populations as shown in online supplementary figure S7. rs11758351 of HIST1H2BF-HIST1H4E did not show a significant association with gout (pmeta=0.40; OR=1.12). rs2285340 of SLC22A12 and rs1165196 of SLC17A1 did not reach a genome-wide level of significance (pmeta=2.47×10−4; OR=1.31; and pmeta=1.28×10−3; OR=1.25, respectively) partly due to statistical fluctuation in relatively small sample sets, although the effects were consistently in the same direction.
Discussion
With clinically defined gout cases, we previously performed a GWAS10 and revealed that ABCG2, SLC2A9, MYL2-CUX2, GCKR and CNIH-2 were associated with gout at a genome-wide significance level (see online supplementary figure S4). A more recent GWAS by Li et al24 with clinically ascertained gout cases revealed three novel loci (BCAS3, RFX3 and KCNQ1) in Han Chinese. In the present study, we performed a gout follow-up study focused on loci not reaching the genome-wide level of significance in the previous GWAS,10 genotyping 1961 SNPs in an additional 1396 cases and 1268 controls. We revealed a total of eight loci to be associated with all gout cases in Japanese males (table 1). Among them, three loci (SLC22A12, SLC17A1 and HIST1H2BF-HIST1H4E) were first identified as gout risk loci at a genome-wide significance level by the present GWAS approach.
Both SLC22A12 and SLC17A1 encode urate transporters at the apical side of the renal proximal tubule16 ,25 (see online supplementary figure S8) and are reportedly associated with SUA level in humans by previous GWASs of SUA.12 ,22 ,26 ,27 Therefore, it is reasonable that SNPs around these loci would display significant associations with gout or sequelae of hyperuricaemia (see also online supplementary note for detail).
The HIST1H2BF and HIST1H4E genes encode histone 1 H2bf and histone 1 H4e, respectively, both of which have a role of binding DNA to form a chromatin structure. Both are replication-dependent histone proteins with expression dependent on cell cycle. Therefore, functional SNPs in this locus might affect the stability of the chromatin structure, varying the cell cycle, cell amount or reaction to inflammation by changing the expression level of histones in the kidney and/or intestine. Since it is also possible that rs11758351 is a surrogate marker near these histone genes, further studies concerning this locus will be necessary.
In this study, we first performed GWASs of gout subtypes, that is, RUE gout and ROL gout (figure 1). From the results of meta-analysis for GWASs of both ROL gout and RUE gout, four shared loci of GCKR, SLC2A9, ABCG2 and CUX2 were identified at a genome-wide significance level, showing the importance of these loci for the pathogenesis of both gout subtypes. Especially for RUE gout, three more loci, SLC22A12, NIPAL1 and FAM35A, were identified to be associated at a genome-wide significance level. As described above, it is compatible for SLC22A12 to be associated with RUE gout, because SLC22A12 (like SLC2A9) encodes a renal urate reabsorption transporter.25 ,28
Of note, NIPAL1 and FAM35A were identified as novel loci by performing GWAS of the RUE gout subtype. Associations with gout and SUA have never been previously reported with NIPAL1 and FAM35A. Furthermore, to our knowledge, there is no study reporting an association between any diseases and NIPAL1 or FAM35A.
NIPAL1, also known as NIPA3, is reportedly expressed on the membrane of some organs including kidney, and to be a magnesium transporter,23 as another magnesium transporter NIPA2.23 Because NIPAL1 was associated with RUE gout (ie, gout with renal urate underexcretion), we hypothesised that NIPAL1 is a urate transporter in the human kidney. However, our functional study did not show urate transport via NIPAL1, regardless of the presence of magnesium (figure 3B). Moreover, localisation to the membrane was not detected for NIPAL1 protein, which was mainly expressed within the distal tubules of human kidney, as revealed by immunohistochemical analysis (figure 4A). A similar result was obtained in confocal microscopic observation, with NIPAL1-expressing oocytes showing intracellular localisation of NIPAL1 protein (see online supplementary figure S6). These findings suggest that NIPAL1 is not a urate transporter and that it might be involved in the indirect regulation of urate transport kinetics. Nevertheless, recent studies have revealed associations between hyperuricaemia and magnesium intake,29 serum magnesium level30 and magnesium excretion.31 Together with previous reports, our findings support the hypothesis that there could be some relationship between gout and magnesium handling via magnesium transporters including NIPAL1, and that the present study could well provide new insights into the genetic background of urate and magnesium handling in patients with gout/hyperuricaemia.
FAM35A is ubiquitously expressed in organs including the kidney, and our immunohistochemical analysis of human kidney also revealed cytosolic immunoreactivity of the FAM35A protein mainly in the distal tubules (figure 4B). Our findings from FAM35A and NIPAL1 suggest the involvement of the distal nephron in gout progression as well as dysfunction in urate handling in humans (see online supplementary figure S9). To date, the molecular function of FAM35A is totally unknown. Although further studies are necessary to confirm this, it is possible that genes near FAM35A including GLUD1 (figure 2E) have some relationship with gout (see also online supplementary note for details).
In addition to studying the Japanese population, we performed a replication study with male Caucasian and NZ Polynesian sample sets for the five newly discovered loci. Since they were not divided into subtypes, further evaluations by meta-analysis were conducted with all gout groups. While other loci were not replicated, rs7903456 of FAM35A was replicated with a significant association with gout (table 2). CUX2, which was reported by both our present and previous gout GWAS in Japanese,10 was also replicated in these sample sets (see online supplementary table S5).
A meta-analysis of all gout with Japanese, Caucasian and NZ Polynesian populations for these five SNPs revealed FAM35A to be associated with all gout at the genome-wide significance level (figure 5B), and that rs2285340 of SLC22A12 and rs1165196 of SLC17A1 showed a significant association but did not reach a genome-wide significance level (see online supplementary figure S7). rs11758351 of HIST1H2BF-HIST1H4E and rs11733284 of NIPAL1 were not associated by this meta-analysis, although these loci showed a genome-wide significant association in the Japanese population. Since this might be due to the differences in LD structure among these populations, a replication analysis with East Asian populations will be necessary for these loci. rs2285340 of SLC22A12 was monomorphic (only G allele) in Caucasians and not associated with NZ Polynesians. Therefore, replication studies of this locus in East Asian populations would also be insightful for future analysis. Although the underlying molecular mechanism of gout by FAM35A is unknown, this locus seems to have a common pathophysiological risk of gout for Japanese, NZ Polynesians and Caucasians.
In summary, we performed GWASs of all gout as well as gout subtypes and identified five loci in addition to the five loci that we reported previously.10 Furthermore, the FAM35A locus showed an association with all gout by meta-analysis among the Japanese, Caucasian and NZ Polynesian sample sets at a genome-wide level of significance. Together with their expression in the renal distal tubules, the identification of NIPAL1 and FAM35A as gout loci suggests the involvement of the distal nephron (see online supplementary figure S9) in the urate handling of the human kidney and in the pathogenesis of gout/hyperuricaemia. These findings could well provide a clue leading to a novel concept for the therapeutic target of gout (see online supplementary figure S10).
Acknowledgments
The authors thank all the participants involved in this study. They are also grateful to members of the BioBank Japan Project for supporting the study. They are indebted to M Watanabe and Y Katsurada (National Defense Medical College) for immunohistochemical analysis; K Gotanda, Y Morimoto, M Miyazawa, T Chiba, Y Utsumi, S Terashige, Y Kato, H Sasaki, Y Takashima, S Tatsukawa, A Akashi, Y Tanahashi, Y Nagao, M Nakajima, H Inoue, S Takeuchi (National Defense Medical College), M Sonoda (Kurume University School of Medicine) and T Tamatsukuri (Jikei University School of Medicine) for genetic analysis; S Ushida (Ikagaku) and H Fujiwara (Midorigaoka Hospital) for Japanese sample collection; R Akuhata, N Aupouri (Ngati Porou Hauora Charitable Trust) and J H Hindmarsh (Research Coordinator, Ngati Porou Hauora Charitable Trust) for NZ Māori (Eastern Polynesian) sample collection from the Rohe (area) of Ngati Porou iwi; Y Oka, S Kanda and C Umatani (the University of Tokyo) for their biomaterial support and technical advice in the oocyte experiment; J Boocock (University of Otago) for eQTL analysis; H Motohashi (Tohoku University), N Hamajima, M Naito (Nagoya University) and H Tanaka (Aichi Cancer Center Research Institute) for helpful discussion.
References
Footnotes
Handling editor Tore K Kvien
AN, HNakaoka, KY, MS, AS and YT contributed equally.
Collaborators Members of the Eurogout Consortium are: Mariano Andres (Sección de Reumatología, Hospital General Universitario de Alicante, Alicante), Leo A Joosten (Department of Internal Medicine and Radboud Institute of Molecular Life Science, Radboud University Medical Center, The Netherlands), Matthijs Janssen (Department of Rheumatology, Rijnstate Hospital, The Netherlands), Tim L Jansen (Department of IQ HealthCare, VieCuri Medical Centre, The Netherlands), Frederic Lioté (INSERM, UMR-S 1132, Hospital Lariboisière, Paris, University Paris Diderot (UFR de Médecine), Sorbonne Paris Cité, Paris), Timothy R Radstake (Department of Rheumatology and Clinical Immunology, Laboratory of Translational Immunology, University Medical Centre Utrecht, The Netherlands, and Department of Immunology, University Medical Centre Utrecht, The Netherlands), Philip L Riches (Rheumatic Diseases Unit, Institute of Genetics and Molecular Medicine, University of Edinburgh, Edinburgh), Alexander So (DAL, Service of Rheumatology, Laboratory of Rheumatology, University of Lausanne, CHUV, Nestlé), Anne-Kathrin Tausche (Department of Rheumatology, University Clinic ‘Carl-Gustav-Carus’, Dresden).
Contributors AN, HNakaoka, KY, MS, AS, YT, TT, NS, TRM and HM conceived and designed this study. YSakurai, HS, II, ATakahashi and MKubo assisted with research design. AN, HNakaoka, MS, YOkada, YKamatani, THigashino, YKawamura, ATokumasu, KO, TK, KW, BS, KP, ATakahashi, MKubo, HOoyama, TS, KIchida and HM collected and analyzed clinical data of Japanese participants. AS, LKS, ND, Eurogout Consortium and TRM collected and analyzed clinical data of replication participants. AN, KY, MS, AS, YShirahama, SS, THigashino, YKawamura, HOgata, MKawaguchi, ID, NS, TRM and HM performed genetic analysis. AN, HNakaoka, MS, AS, YOkada, YKamatani, TN, HNakashima, ATakahashi, TRM and HM performed statistical analysis. AN, YT, TT, KInoue, TYasujima, HY, HS and HM performed functional analysis and localization analysis. AN, HNakaoka, KY, MS, AS, YT, YOkada, YKamatani, TN, TT, KInoue, TYasujima, HY, YOhkawa, NS, TRM and HM analyzed data. ID, TI, MH, SF, TYokoo, THosoya, KIchida provided intellectual input and assisted with the preparation of the manuscript. AN, HNakaoka, KY, MS, AS, YT, NS, TRM and HM wrote the manuscript.
Funding This study was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan, including MEXT KAKENHI (Nos. 25293145 and 15K15227), Grants-in-Aid for Scientific Research on Priority Areas (No. 17015018) and Innovative Areas (Nos. 221S0001 and 221S0002) and a JSPS KAKENHI Grant (Nos. 16H06277 and 16H06279), the Ministry of Health, Labour and Welfare of Japan, the Ministry of Defense of Japan, the Japan Society for the Promotion of Science, the Kawano Masanori Memorial Foundation for Promotion of Pediatrics, the Gout Research Foundation of Japan and the Health Research Council of New Zealand. The BioBank Japan Project was supported by MEXT of Japan.
Competing interests TT, KIchida, NS and HM have a patent pending based on the work reported in this paper.
Patient consent Obtained.
Ethics approval This study was approved by the institutional ethical committees, and written consent was obtained from all of its participants. All involved procedures were performed in accordance with the Declaration of Helsinki.
Provenance and peer review Not commissioned; externally peer reviewed.