Article Text

Abstract
Objectives TNFSF4 (encodes OX40L) is a susceptibility locus for systemic lupus erythematosus (SLE). Risk alleles increase TNFSF4 expression in cell lines, but the mechanism linking this effect to disease is unclear, and the OX40L-expressing cell types mediating the risk are not clearly established. Blockade of OX40L has been demonstrated to reduce disease severity in several models of autoimmunity, but not in SLE. We sought to investigate its potential therapeutic role in lupus.
Methods We used a conditional knockout mouse system to investigate the function of OX40L on B and T lymphocytes in systemic autoimmunity.
Results Physiologically, OX40L on both B and T cells contributed to the humoral immune response, but B cell OX40L supported the secondary humoral response and antibody affinity maturation. Our data also indicated that loss of B cell OX40L impeded the generation of splenic T follicular helper cells. We further show that in two models of SLE—a spontaneous congenic model and the H2-IAbm12 graft-versus-host-induced model—loss of B cell OX40L ameliorates the autoimmune phenotype. This improvement was, in each case, accompanied by a decline in T follicular helper cell numbers. Importantly, the germline knockout did not exhibit a markedly different phenotype from the B cell knockout in these models.
Conclusions These findings contribute to a model in which genetically determined increased OX40L expression promotes human SLE by several mechanisms, contingent on its cellular expression. The improvement in pathology in two models of systemic autoimmunity indicates that OX40L is an excellent therapeutic target in SLE.
- systemic lupus erythematosus
- B cells
- OX40L
- T follicular helper cells
- autoantibodies
This is an Open Access article distributed in accordance with the terms of the Creative Commons Attribution (CC BY 4.0) license, which permits others to distribute, remix, adapt and build upon this work, for commercial use, provided the original work is properly cited. See: http://creativecommons.org/licenses/by/4.0/
Statistics from Altmetric.com
Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterised by autoantibodies against nuclear antigens along with the deposition of immune complexes.1 2 As with most other autoimmune diseases, environmental and genetics factors contribute to the risk of developing SLE. Genome-wide association studies have revealed over 50 susceptibility loci.3 4 TNFSF4 (tumour necrosis factor ligand family, member 4, CD252) is an established susceptibility gene for SLE4 5 and for several other autoimmune diseases.6–9 Fine-mapping of this locus in SLE identified two independent association signals upstream of TNFSF4 in multiple ancestries.10 These two signals align with separate expression quantitative trait loci, each one associated with elevated expression of TNFSF4 in Epstein Barr virus (EBV) lymphoblastoid cell lines,11 suggesting that TNFSF4 transcription is upregulated in individuals harbouring risk alleles.
TNFSF4 encodes the costimulatory molecule, OX40L, a type II transmembrane protein expressed on several immune cell types on activation, including anitigen presenting cells (APCs), such as dendritic cells (DCs), B cells and macrophages,12–14 activated T cells,15 16 and mast cells and vascular endothelial cells.17 In contrast, its only known receptor, OX40, is expressed mainly on activated CD4+ T cells.18–21 The OX40L-OX40 signalling pathway is fundamental for effector T cell proliferation and memory T cell development, maintenance of cytokine production by T cells and DCs, increasing Ig production, and promoting plasma cell development.15 22–27 Nevertheless, how these various functions relate to the cell types expressing OX40L is still unclear. Constitutive expression of OX40L on T cells has been shown to induce spontaneous autoimmunity in C57BL/6 mice.23 A recent study showed that OX40L expression on a subset of myeloid DCs is implicated in the pathogenesis of SLE.28 The beneficial effect of blocking the OX40L-OX40 signalling pathway has been shown in several different mouse models of autoimmune diseases,17 but experimental evidence of its efficacy in SLE is unknown.
We sought to understand the function of OX40L using CD4+ T cell and B cell conditional knockout mice. We investigated the role of OX40L using immunisation and we went on to determine how the loss of OX40L affected the pathology in two different SLE mouse models.
Materials and methods
Mice
A bacterial artificial chromosome (BAC) clone encoding the extracellular domain and 3′-untranslated region of Tnfsf4 was obtained from a C57BL/6-derived genomic library. The Tnfsf4 conditional targeting vector was constructed using recombineering,29 as described in online supplementary figure S1A. The mice (Tnfsf4fl/fl) were made according to a standard gene targeting approach in A9 embryonic stem cells (ES). We used (129xC57BL/6)F1 ES; therefore, microsatellite analyses were undertaken to confirm that the targeting vector had recombined on the C57BL/6 chromosome. The mice were backcrossed for eight generations on the C57BL/6 background. Tnfsf4fl/fl mice were crossed with β-actin-cre, CD4-cre and CD19-cre (Jackson Laboratories) to obtain Tnfsf4−/−, Tnfsf4fl/fl/CD4-cre and Tnfsf4fl/fl/CD19-cre, respectively. Before each experiment mice were genotyped by PCR. The primers and expected PCR product size are listed in supplementary table S1. B6.Sle16 mice were bred in-house and B6.Sle16.Tnfsf4−/− were generated by crossing them with Tnfsf4−/− mice. B6-H2bm12 mice were purchased from the Jackson Laboratories (B6(C).H2-Ab1bm12/KhEgJ; strain no. 001162, https://www.jax.org/strain/001162). The mice used were female, 8–12 weeks old and housed in specific pathogen-free conditions. All animal procedures were performed in accordance with institutional guidelines and approved by the UK Home Office.
Supplementary file 1
In vitro analysis of OX40L expression
To assess OX40L expression in vitro, different cell subsets were purified from mouse spleen using LS Columns and MACS Technology (Miltenyi Biotec) and stimulated as described before.16 24 Briefly, single cell suspensions were obtained from collagenase-treated spleens, and B cells, DCs and T cells were then purified incubating the splenic cell suspension with anti-CD43(Ly-48) microbeads, anti-CD11c microbeads or CD4+ T cell isolation kit, respectively, following the manufacturers’ protocols. The purity of each subpopulation was tested routinely by fluorescence-activated cell sorting (FACS) and a value >95% was measured for each purification. Purified B cells and DCs were stimulated for 72 hours with anti-CD40 (Clone3/23 at 2.5 µg/mL) plus F(ab’)2 anti-mouse IgM (10 µg/mL) or anti-CD40 alone, respectively. T cells were stimulated with anti-CD3 (0.005 µg/mL), IL-2 (100 U/mL) and IL-12 (10 ng/mL) for 7 days. After stimulation, the cells were harvested and analysed by FACS for OX40L expression.
Flow cytometry
Flow cytometry was performed using a five-colour or six-colour staining protocol and analysed with a BD FACSVerse (BD Biosciences, San Jose, California, USA). The following Abs were used: anti-CD4 (GK.5), anti-CXCR5 (L138D7), anti-PD1(29F.1a12), anti-CD62L (MEL-14), anti-CD44 (IM7), anti-B220 (RA3-6B2), anti-GL7 (GL7), anti-CD138 (281-2) and anti-IgD (11–26 c.2a). Abs were purchased from BioLegend (San Diego, California, USA). Staining was performed in the presence of a saturating concentration of 2.4G2 mAb (anti-FcγRII/III). Data were analysed using FlowJo V.9 (Tree Star, Ashland, Oregon, USA).
Immunisation and ELISA
Mice were immunised subcutaneously with 50 μg 4-hydroxy-3-nitrophenylacetyl-chicken gamma globulin (NP-CGG) in complete Freund’s adjuvant. For the analysis of the secondary response mice were reimmunised with 50 µg of NP-CGG in incomplete Freund’s adjuvant 35 days after receiving the first immunisation. Serum was collected on days 7, 14, 28 and 42, and titres of isotype-specific low-affinity and high-affinity antibodies to NP were measured by ELISA in plates coated with either NP25-BSA or NP4-BSA (4-hydroxy-3-nitrophenylacetyl hapten conjugated to bovine serum albumin), respectively.30 Briefly NUNC plates were coated with the antigen at 5 µg/mL in borate buffered saline (BBS) overnight at 4°C. Plates were washed with phosphate buffered saline (PBS) and then blocked for 1 hour at room temperature with 0.5% BSA in PBS. Samples were diluted in dilution buffer (PBS 2%, bovine serum albumin (BSA) 0.05% Tween-20) and added, in duplicate, to the plates for 3 hours at 37°C. Plates were washed and incubated with alkaline phosphatase (AP)-conjugated secondary antibody specific for the different Ig isotype (SouthernBiotech) for 3 hours at room temperature. Plates were developed with p-nitrophenylphosphate (Sigma). A standard serum was generated from a pool of reactive serum of immunised wild-type mice. Absorbance was read at 405 nm and data were expressed as arbitrary ELISA unit (AEU) in reference to a standard curve obtained by serial dilution of the standard serum.
cGvHD mouse model and autoantibody assays
Knockout and control mice were injected intraperitoneally with 5×107 splenocytes from B6.H2bm12 mice. Briefly, splenocytes were obtained as a single cell suspension by mashing the spleen collected through 70 µm cell strainers using the plunger from a syringe. After lysis of the red blood cells, splenocytes were counted and resuspended at 5×108 cells/mL in PBS and 100 µL was injected in each mouse. Serum was collected on days 14, 28 and 42, and titres of IgG antibodies to double-stranded deoxyribonucleic acid (dsDNA) were measured by ELISA using dsDNA (100 µg/mL) or single-stranded deoxyribonucleic acid (ssDNA) (10 µg/mL) in BBS buffer as coating antigen. Bound Abs were detected with AP-conjugated goat anti-mouse IgG (-chain specific) (Sigma-Aldrich) or IgM (Southern Biotechnology Associates). The results were expressed as AEU relative to a standard positive sample derived from an MRL/Mplpr/ lpr mice pool.
Total serum IgG and IgM levels
Total serum IgM and IgG levels were assayed by capture ELISA as previously described.31
IgG, IgM and C3 kidney deposition
Fluorescein (FITC)-conjugated goat Abs against mouse total IgG (1/400 dilution; Sigma-Aldrich), mouse total IgM (1/200 dilution, eBioscience) and against mouse C3 (1/50 dilution; ICN Pharmaceuticals) were used on snap-frozen kidney sections. The staining with FITC-conjugated Abs was quantified as previously described31 and expressed as arbitrary fluorescence units.
Statistical analysis
Where appropriate either the Student’s t-test, two-way analysis of variance (ANOVA) or one-way ANOVA followed by Fisher’s least significant difference (LSD) multiple comparison test was performed using GraphPad Prism V.6.00 for Windows (GraphPad Software, La Jolla, California, USA).
Results
Generation of Tnfsf4 conditional knockout strains
We generated a floxed Tnfsf4 mouse (Tnfsf4fl/fl) on the C57BL/6 genetic background (see online supplementary figure S1A,B) to avoid the confounding effects caused by epistatic interactions between 129 and C57BL/6 genes that promote an autoimmune phenotype.32 Germline knockout (KO) mice were obtained by crossing Tnfsf4fl/fl with the β-actin Actb-cre mouse strain. Conditional T cell Tnfsf4fl/fl(CD4)−/− and B cell Tnfsf4fl/fl(CD19)−/− specific knockout mice were created by crossing with CD4-cre33 and CD19-cre mice,34 respectively. Lack of OX40L was observed in all cell types from Tnfsf4−/− mice, while a cell-specific deletion was confirmed in the conditional knockouts (see online supplementary figure S1C).
B cell OX40L promotes antibody affinity maturation
Conflicting data have been reported on the importance of the OX40L-OX40 pathway in controlling T-dependent antibody responses.24 26 35 Thus, we explored this response by immunising the three KO strains and a control group with NP-CGG, a well-studied T cell-dependent antigen. All three strains showed significantly lower titres of low-affinity IgG2a and IgG2b antibodies against NP25-BSA compared with wild-type mice (figure 1A). In contrast, the IgG1 response was hardly affected by the lack of OX40L. Affinity maturation during the primary response was also assessed by measuring antibody against NP4-BSA on day 28 and by calculating the affinity maturation index (ratio of high-affinity to low-affinity antibody responses). All three knockout strains displayed lower titres of high-affinity IgG2a and IgG2b (figure 1B,C) compared with wild-type animals and a lower affinity maturation index (figure 1D), which suggested OX40L contribution in the antibody affinity maturation process. To investigate the role of OX40L in the secondary immune response, mice were then boosted with NP-CGG on day 35, and the high-affinity antibody response was measured 1 week later. Tnfsf4−/− and Tnfsf4fl/fl(CD19)−/− mice both showed a significantly impaired IgG2a and IgG2b memory response compared with control mice, associated with a lower affinity maturation index (figure 1C,E). In contrast, the memory response in the Tnfsf4fl/fl(CD4)−/− mice was normal (figure 1C). These results indicate a role for both B and T cell OX40L in the primary immune response, with a distinct role for B cell OX40L in the affinity maturation of the secondary humoral immune response.

OX40L in T cell-dependent humoral response. Wild-type controls, Tnfsf4−/−, Tnfsf4fl/fl(CD19)−/− and Tnfsf4fl/fl(CD4)−/− mice were immunised with NP-CGG in CFA and reimmunised on day 35 with NP-CGG in IFA. Sera were collected on days 7, 14 and 28 for the primary response and on day 42 for the secondary response. (A) Titres of NP-specific low-affinity antibody measured with NP25-BSA. (B) Titres of NP-specific high-affinity antibodies on day 28 measured with NP4-BSA. (C) Titres of NP-specific high-affinity antibodies on day 42 measured with NP4-BSA. (D) Affinity maturation index calculated as ratio of the titres of IgG detected with NP4-BSA to those with NP25-BSA on day 28. (E) Affinity maturation index calculated as the ratio of the titres of IgG detected with NP4-BSA to those with NP25-BSA on day 42. Each symbol represents an individual mouse; dots in (A) and bars in (B–E) indicate the mean titre, each being shown mean±SEM. AEU is arbitrary ELISA unit; N.S. is not significant; *p<0.05, **p<0.01 and ***p<0.001 (A, two-way ANOVA; B–E, one-way ANOVA). ANOVA, analysis of variance; CFA, complete Freund’s adjuvant; IFA, incomplete Freund’s adjuvant; NP-CGG, nitrophenylacetyl-chicken gamma globulin; NP25-BSA, NP4-BSA, 4-Hydroxy-3-nitrophenylacetyl hapten conjugated to bovine serum albumin.
OX40L is essential for T cell activation
As the impaired humoral response could be a consequence of defective T cell activation, we decided to investigate the splenic T cell composition (see online supplementary figure S2A) of immunised mice on days 14 and 42 (figure 2). By day 14, Tnfsf4−/− and Tnfsf4fl/fl(CD4)−/− had a markedly lower proportion of effector T and effector/memory CD4+ T cells. In contrast, Tnfsf4fl/fl(CD19)−/− mice showed only a reduction in the proportion of effector/memory T cells, indicating that B cell OX40L may not play a major role in priming naïve T cells. On day 42, all three knockout strains had fewer T effector cells than control mice (figure 2). Interestingly, Tnfsf4−/− and Tnfsf4fl/fl(CD19)−/− mice also showed a small, but statistically significant, increment in the frequency of central memory T cells, suggesting that OX40L may regulate the balance between effector and central memory T cells during the recall response (figure 2). Our data confirmed the previous reported role of OX40-OX40L signalling in T cell activation and development of T effector memory cells.18 22 23 36 The explanation for the difference in the secondary humoral response between Tnfsf4fl/fl(CD4)−/− and Tnfsf4fl/fl(CD19)−/− mice was not evident. We therefore decide to investigate further analysing the extent of the germinal centre (GC) reaction in the immunised mice.
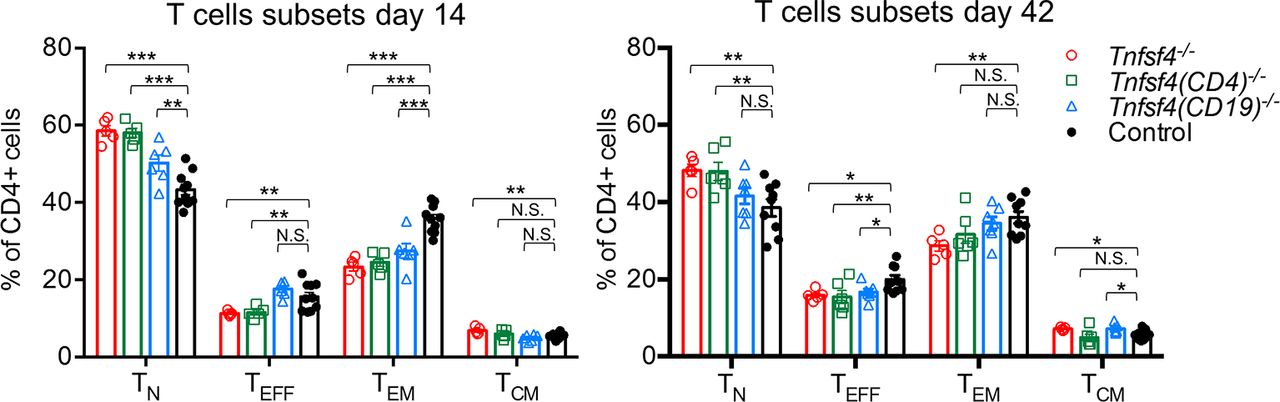
Role of OX40L in T cell activation. Wild-type controls, Tnfsf4−/−, Tnfsf4(CD19)−/− and Tnfsf4(CD4)−/− mice were immunised with NP-CGG in CFA and reimmunised on day 35 with NP-CGG in IFA. Spleens were taken and analysed by FACS either on day 14 or day 42. Quantification of naïve (CD4+, CD62L+, CD44low) T cells, effector (CD4+, CD62Llow, CD44low) T cells, effector/memory (CD4+, CD62Llow/neg, CD44hi) T cells and central/memory (CD4+, CD62L+, CD44hi) T cells on day 14 (left) and day 42 (right). Each symbol represents an individual mouse. Bars indicate the mean±SEM. N.S., not significant; *p<0.05, **p<0.01 and ***p<0.001 (one-way analysis of variance). CFA, complete Freund’s adjuvant; FACS, fluorescence-activated cell sorting; IFA, incomplete Freund’s adjuvant; NP-CGG, nitrophenylacetyl-chicken gamma globulin.
OX40L on B cells supports plasma cell development
All three groups of immunised KO mice showed no difference in the GC B cell population (see online supplementary figure S2B) on day 14 (figure 3A), although during the secondary response, 1 week after the rechallenge, Tnfsf4−/− mice showed a smaller proportion of GC B cells (figure 3A). Similarly, no differences were detected in the plasma cell frequency on day 14, but Tnfsf4−/− and Tnfsf4fl/fl(CD19)−/− mice showed a significantly lower percentage of plasma cells on day 42 (figure 3B).

OX40L function in GC reaction. Wild-type controls, Tnfsf4−/−, Tnfsf4(CD19)−/− and Tnfsf4(CD4)−/− mice were immunised with NP-CGG in CFA and reimmunised on day 35 with NP-CGG in IFA. Spleens were taken and analysed by FACS either on day 14 or day 42. (A) Frequency of GC B cell (B220+, GL7+, IgD−) presented as frequency among the B220+ population. (B) Percentage of plasma cells (B220low, CD138hi). (C) Gating of T follicular helper (TFH) (CD4+, CXCR5+, PD-1hi) and pre-T follicular helper (pre-TFH) (CD4+, CXCR5+, PD-1Low/neg) cells. (D) PD-1 expression level in CD4+ cells assessed by FACS. (E) Frequency of CXCR5+ cells presented as frequency among the CD4+ population. (F) Quantification of pre-GC TFH and (G) GC-TFH cells as gated in (C) presented as frequency among the CD4+ population. Each symbol represents an individual mouse. Bars indicate the mean±SEM, N.S., not significant; *p<0.05, **p<0.01 and ***p<0.001 (one-way analysis of variance). CFA, complete Freund’s adjuvant; FACS, fluorescence-activated cell sorting; GC, germinal centre; IFA, incomplete Freund’s adjuvant; NP-CGG, nitrophenylacetyl-chicken gamma globulin.
B cell OX40L is essential for TFH maturation
Having demonstrated the importance of OX40L in T cell activation and in plasma cell development, we investigated its possible role in T follicular helper cell (TFH) maturation. We identified the GC TFH population as a subset of CD4+ T cells expressing CXCR5 and high levels of PD-1 (CXCR5+PD-1hi) (figure 3C,F), and in figure 3G the frequencies of splenic GC TFH cells (as a proportion of CD4+ T cells) following immunisation are illustrated. There were fewer GC TFH cells in the spleens of Tnfsf4−/− and Tnfsf4fl/fl(CD19)−/− compared with wild-type mice during both the primary and secondary responses. In contrast, no differences were observed between controls and Tnfsf4fl/fl(CD4)−/− mice. Both Tnfsf4−/− and Tnfsf4fl/fl(CD19)−/− mice showed a reduction in the expression levels of PD1 at both time points, and importantly displayed a greater frequency of CXCR5+ PD1low cells (TFH precursors) in the CD4+ population compared with control mice on day 42 (figure 3D,F). Interestingly, Tnfsf4−/− and Tnfsf4fl/fl(CD19)−/− mice also revealed a reduced number of CXCR5+ cells during the primary (day 14) but not the secondary response (day 42) (figure 3E).
Lack of OX40L reduces TFH number and ameliorates the lupus phenotype
In view of the genetic association of TNFSF4 and SLE and the functional results outlined above, we investigated the effect of loss of OX40L in SLE using two different mouse models: a congenic model and a graft-versus-host model.
Tnfsf4−/− mice were crossed with B6.Sle16 lupus-prone mice, which are characterised by development of humoral autoimmunity associated with splenomegaly, high level of total IgG and IgM, autoantibodies production and glomerulonephritis linked to Ig and C3 deposition in the kidney.31 32 The resultant B6.Sle16.Tnfsf4−/− female animals were monitored for 9 months (figure 4). The absence of OX40L was associated with a marked reduction in splenomegaly (figure 4A,B) and a lower serum level of total IgG and IgM (figure 4C). No detectable levels of IgG anti-DNA were observed either in the knockout or the B6.Sle16 control group. However, when we analysed IgM anti-ssDNA autoantibodies, a significant lower titre was observed in mice lacking OX40L compared with the B6.Sle16 group (figure 4D). To investigate the effect of loss of OX40L on target organs, we quantified glomerular IgG, IgM and complement C3. As expected fluorescent quantification revealed significantly lower amount of IgG and IgM deposition in the glomerular in the absence of OX40L; in contrast a similar level of C3 deposition was observed (see online supplementary figure S3). Mice lacking OX40L had less T cell activation and higher proportions of central memory and naïve T cells (CD62L+ CD44hi cells) (figure 4E). Consistent with the immune response data (figure 3), the B6.Sle16.Tnfsf4−/− showed a fivefold reduction, relative to the B6.Sle16 mice, in the proportion of CD4+ TFH cells (figure 4F), along with a dramatic reduction of PD-1 expression on CD4+ cells (figure 4G). Furthermore, the percentage of plasma cells and GC B cells (B220+ IgD GL7+) was also significantly lower in the absence of OX40L (figure 4H,I).

OX40L deficiency ameliorates the phenotype of B6.Sle16 lupus-prone mice. Comparison between female B6.Sle16 and B6.Sle16.Tnfsf4−/− female mice at 9 months of age. (A) Quantitation of spleen/body weight ratio and spleen weight. (B) Absolute number of cells per spleen. (C) Serum level of IgG and IgM at 6 and 9 months. (D) Titre of IgM anti-dsDNA and anti-ssDNA. (E) Quantitation of naïve (TN) (CD4+, CD62L+, CD44low), (TEFF) effector (CD4+, CD62Llow, CD44low), TEM effector/memory (CD4+, CD62Llow/neg, CD44hi) and TCM central/memory (CD4+, CD62L+, CD44hi) T cells expressed as a percentage of CD4+ cells and absolute number. (F) GC TFH cells (CD4+, CXCR5+, PD-1hi) presented as frequency among the CD4+ population and absolute number. (G) PD-1 expression level in CD4+ cells assessed by FACS. (H) GC B cell (B220+, GL7+, IgD−) presented as frequency among the B220+ population and absolute number. (I) Percentage and absolute number of plasma cells (B220low, CD138hi). Each symbol represents an individual mouse. Bars indicate the mean±SEM N.S., not significant; *p<0.05, **p<0.01 and ***p<0.001 (t-test). dsDNA, double-stranded deoxyribonucleic acid; FACS, fluorescence-activated cell sorting; GC, germinal centre; ssDNA, single-stranded deoxyribonucleic acid.
We then used the I-Abm12 chronic graft-versus-host-disease (cGvHD) mouse model, in which an allogeneic interaction of T and B cells expressing different major histocompatibility complex (MHC) class II (I-A) induces an SLE-like phenotype.37 38 As shown in figure 5A, Tnfsf4−/− and Tnfsf4fl/fl(CD19)−/− mice injected with B6.H2-Ab1bm12 splenocytes developed a lower titre of IgG anti-dsDNA compared with controls. In addition, both knockout groups showed a lower percentage of effector/memory T and TFH cells (figure 5B,C). A trend towards a lower percentage of plasma and GC B cells was observed in both OX40L-deficient groups (figure 5D,E). Of note, no differences were seen between Tnfsf4−/− and Tnfsf4fl/fl(CD19)−/− mice, indicating that the observed differences are primarily due to the lack of OX40L on B cells.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
OX40L deficiency diminishes anti-dsDNA antibody production in the cGvHD model. Female wild-type controls, Tnfsf4−/− and Tnfsf4(CD19)−/− mice were injected intraperitoneally with 5×107 splenocytes from B6.H2bm12 female mice. Sera were collected on days 14, 28, 42 and 56. On day 56, spleens were collected and analysed by FACS. (A) Titre of IgG anti-dsDNA in the sera of injected mice at different time points. (B) Quantification of naïve (TN) (CD4+, CD62L+, CD44low), (TEFF) effector (CD4+, CD62Llow, CD44low), (TEM) effector/memory (CD4+, CD62Llow/neg, CD44hi) and (TCM) central/memory (CD4+, CD62L+, CD44hi) T cells expressed as percentage of CD4+ cells. (C) Quantification of GC TFH cells (CD4+, CXCR5+, PD-1hi) presented as frequency among the CD4+ population. (D) Frequency of GC B cells (B220+,GL7+, IgD−) presented as frequency among the B220+ population. (E) Percentage of plasma cells (B220low, CD138hi). Each symbol represents data from an individual mouse. Bars indicate the mean±SEM. N.S., not significant; *p<0.05, **p<0.01 and ***p<0.001 (one-way analysis of variance). cGvHD, chronic graft-versus-host-disease; dsDNA, double-stranded deoxyribonucleic acid; FACS, fluorescence-activated cell sorting; GC, germinal centre.
Discussion
The TNFSF4 locus (that encodes OX40L) shows association with several autoimmune diseases; it has one of the most consistent and strongest genetic risk factors in SLE. OX40L has a well-established role in the activation and maintenance of T cell-mediated immune responses. However, the diversity of cells that express OX40L is such that a pathogenic mechanism relating the genetic findings to disease has not been clearly established. In this study, we generated B and CD4+ T cell OX40L conditional knockout mice, alongside a complete OX40L knockout, to explore and compare the function of OX40L on these cells.
Although a role for OX40L in the T-dependent antibody response has been suggested, conflicting results using different OX40L-deficient mice have been reported.24 35 These contradictory results may be partly explained by variability in genetic background.39 Our conditional knockout mice were on a pure C57BL/6 background and, in accord with the one study,24 our Tnfsf4−/− mice showed a reduced primary and secondary antibody response. However, while the Tnfsf4fl/fl(CD19)−/− mice showed the same phenotype as the Tnfsf4−/− mice, the Tnfsf4fl/fl(CD4)−/− mice had a normal secondary response, indicating that only OX40L expression by B cells is essential for the generation of an effective secondary humoral response and by implication B cell memory. We then investigated whether this defective humoral response was due to impaired T cell activation; as expected, Tnfsf4−/− mice showed lower percentage of T effector and T effector memory cells (figure 2). The same defect, although at a lower extent, was also shown by both conditional knockouts, despite the normal secondary response in Tnfsf4fl/fl(CD4)−/− mice. These results suggest that B cell OX40L may be involved in biological processes that promote memory responses that are independent of T cell activation.
T cell-dependent B cell immune response involves both an extrafollicular response, which generates short-lived plasma cells and an early wave of low-affinity antibody production, and a GC response, which gives rise to long-lived plasma or memory cells and a later wave of high-affinity antibodies. OX40L has been previously suggested to be essential for the development of high-affinity Ig-producing plasma cells26; however, no further evidence has been subsequently reported. In our study, alongside an impaired memory response (figure 1C), there were fewer plasma cells on day 42 in Tnfsf4−/− and Tnfsf4fl/fl(CD19)−/− mice (figure 3B), which suggests that B cell OX40L contributes to an effective GC reaction.
We show that Tnfsf4−/− and Tnfsf4fl/fl(CD19)−/− mice have a lower percentage of GC TFH, one of the main contributors to the GC reaction. The development of mature GC TFH, which characteristically expresses CXCR5, along with high levels of the surface receptors ICOS, CD40 ligand (CD40L), PD-1 and importantly OX40,40 41 includes two stages: after activation, a fraction of CD4+ T cells migrate towards B cell follicles by upregulating the chemokine receptor CXCR5, and these TFH precursors then interact with antigen-presenting B cells at the border of the B cell follicle and T cell zone and fully maturate into functional GC TFH cells.41 In particular OX40L has been shown to be essential for the expression of CXCR5 and the consequent migration of T cells at the T/B border of B cell follicles,22 42 43 providing the first evidence of the role of OX40L in this process. Our results corroborate this finding; we found that fewer CXCR5+ T cells were generated during the primary response in Tnfsf4−/− and Tnfsf4fl/fl(CD19)−/− mice (figure 3E). Whether OX40L-OX40 signal is responsible for the induction, maturation or maintenance of TFH cells and which cell types expressing OX40L are necessary is still unclear; however, a recent work by Tahiliani and colleagues44 shows a markedly diminished humoral response and production of fewer TFH cells in OX40 KO mice following immunisation with vaccinia virus. In particular the authors show a direct association between OX40+ TFH cells and OX40L-expressing DCs and B cells at the T/B borders and GC providing supportive evidence to how a sustained OX40L-OX40 signal on TFH cells is necessary for the induction of TFH cells and their maturation to maintain a proper GC reaction. In our study, the reduced numbers of TFH cells in Tnfsf4−/− and Tnfsf4fl/fl(CD19)−/− mice were accompanied by an increase in CXCR5+ PD1low cells during the secondary response (figure 3F,G). Since low levels of cell-surface PD1 have been shown to characterise TFH precursor cells,45 our data suggest a novel role for OX40L on B cells: after activation by DCs, immature TFH cells migrate towards the T/B borders of the B cells follicles, where activated antigen presenting B cells induced their maturation into the GC TFH resident state and their maintenance by sustaining OX40L-OX40 signalling.
TNFSF4 has been reproducibly associated with SLE.4 5 A recent important study from Jacquemin and colleagues28 demonstrated that stimulation through OX40 induced T cells to express TFH cells-specific genes such as Bcl6 and CXCR5. They also observed a positive correlation between disease activity, percentage of blood TFH cells and frequency of OX40L+myeloid APC, suggesting OX40L-OX40 axis as a contributor factor in the aberrant TFH response observed in SLE.46 47 However, the ability to study tissue TFH in humans is limited. In our study, although in a murine model, the generation of TFH cells in the spleen is similarly impeded in the B cell conditional knockout and in the germline Tnfsf4 knockout, indicating the importance of B cell OX40L. In the human study,28 there was no correlation between blood B cells expressing OX40L and TFH cells. However, this lack of correlation could be a consequence of the compartmentalisation of activated B cells expressing OX40L in the secondary lymphoid organs rather than an evidence of their lack of involvement in the development of pathogenic TFH cells in SLE.
In our study, to elucidate the role of OX40L in SLE, we used two different SLE mouse models, and in particular the GvHD model was chosen to investigate the role of OX40L on B cells during the B–T cell interaction. In both models of systemic autoimmunity, the lack of OX40L-OX40 signalling was associated with amelioration of the disease phenotype, as shown by a reduced production of anti-dsDNA autoantibodies and Ig kidney deposition together with reduced numbers of GC TFH (figures 4F and 5C) and plasma cells (figures 4I and 5E). These data suggest that OX40L supports the expression of the disease phenotype as well as autoantibody production. This conclusion is further strengthened by the observation that blockade of OX40L reduces degree of proteinuria associated with glomerulonephritis in an accelerated murine model.48
The results presented in this paper support a mechanism by which genetically determined elevated expression of OX40L predisposes to SLE via increased B cell expression, which in turn supports TFH development. In light of the argument that genetic factors augment the likelihood of success with a drug target,49 our data strongly support exploration of this therapeutic strategy. It is potentially important for optimal treatment to know which OX40L-expressing cell types should be targeted, and the defined risk alleles at TNFSF4 further raise the possibility that genetic screening may identify individuals most likely to benefit from OX40L inhibition.
Acknowledgments
We thank Liliane Fossati-Jimack, Marta Szajna and Christopher L Pinder for their technical support.
References
Footnotes
Handling editor Tore K Kvien
Contributors AC designed, performed and analysed experiments and wrote the manuscript. UE performed experiments, helped with the statistical analysis and discussed the data. THM performed experiments. DSCG discussed the data and edited the manuscript. MB and TJV designed experiments, discussed the data and wrote the manuscript.
Funding This work was financed by the Wellcome Trust programme grant 17966/Z/2008. DSCG and TJV were awarded an Arthritis Research UK Project Grant (20265).
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement There are no additional unpublished data. The mouse model described in this study is available to other researchers on request and has already been shared with other investigators.
Correction notice This article has been corrected since it published Online First. The fourth author’s name has been corrected to Deborah S Cunninghame Graham.