Article Text

Abstract
Objective The zinc-ZIP8-MTF1 axis induces metallothionein (MT) expression and is a catabolic regulator of experimental osteoarthritis (OA) in mice. The main aim of the current study was to explore the roles and underlying molecular mechanisms of MTs in OA pathogenesis.
Methods Experimental OA in mice was induced by destabilisation of the medial meniscus or intra-articular injection of adenovirus carrying a target gene (Ad-Zip8, Ad-Mtf1, Ad-Epas1, Ad-Nampt, Ad-Mt1 or Ad-Mt2) into wild type, Zip8fl/fl; Col2a1-Cre, Mtf1fl/fl; Col2a1-Cre and Mt1/Mt2 double knockout mice. Primary cultured mouse chondrocytes were infected with Ad-Mt1 or Ad-Mt2, and gene expression profiles analysed via microarray and reverse transcription-PCR. Proteins in human and mouse OA cartilage were identified via immunostaining. Chondrocyte apoptosis in OA cartilage was determined using terminal deoxynucleotidyl transferase (TdT)-mediated deoxyuridine triphosphate (dUTP) nick end labelling (TUNEL).
Results MTs were highly expressed in human and mouse OA cartilage. Hypoxia-inducible factor 2α, nicotinamide phosphoribosyltransferase and several proinflammatory cytokine pathways, as well as the zinc-ZIP8-MTF1 axis were identified as upstream regulators of MT expression. Genetic deletion of Mt1 and Mt2 enhanced cartilage destruction through increasing chondrocyte apoptosis. Unexpectedly, aberrant overexpression of MT2, but not MT1, induced upregulation of matrix-degrading enzymes and downregulation of matrix molecules through nuclear factor-kappa B (NF-κB) and activator protein-1 (AP-1) activation, ultimately leading to OA.
Conclusions MTs play an antiapoptotic role in post-traumatic OA. However, aberrant and chronic upregulation of MT2 triggers an imbalance between chondrocyte anabolism and catabolism, consequently accelerating OA development. Our findings collectively highlight pleiotropic roles of MTs as regulators of chondrocyte apoptosis as well as catabolic and anabolic pathways during OA pathogenesis.
- Osteoarthritis
- Chondrocytes
- Arthritis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Osteoarthritis (OA) is characterised by cartilage destruction, synovial inflammation, osteophyte formation and subchondral bone sclerosis.1–3 Pathogenic mechanisms of OA include increased production of matrix-degrading enzymes, such as matrix metalloproteinases (MMPs) and a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTS), cessation of cartilage-specific extracellular matrix (ECM) synthesis, and chondrocyte apoptosis.4–6 We recently demonstrated that the zinc-ZIP8-MTF1 axis is a catabolic regulator of experimental OA pathogenesis in mice.7 Upregulation of the Zn2+ importer, ZIP8, elevates intracellular Zn2+ levels in OA cartilage. Zn2+ influx through ZIP8 activates metal regulatory transcription factor-1 (MTF1), in turn, enhancing expression of matrix-degrading enzymes and OA cartilage destruction.7
Metallothioneins (MTs) are known targets of MTF1.8–10 MTs regulate metal homoeostasis by virtue of their specific binding to metal ions, such as Zn2+. Moreover, MTs are involved in transferring bound Zn2+ ions to zinc-dependent enzymes and transcription factors, leading to alterations in enzymatic activity and DNA binding affinity, respectively.11 ,12 Various stimuli, such as heavy metals, oxidative stress and hypoxia, induce MT expression,13–16 and upregulation of MTs has been reported in OA cartilage.17 However, their specific roles in OA pathogenesis remain largely unknown. Our group previously showed enhanced OA cartilage destruction in Mt1/Mt2 double knockout (KO) mice, suggesting that a negative feedback loop maintains Zn2+ homoeostasis during zinc-ZIP8-MTF1 axis-induced OA pathogenesis.7
In this study, we investigated the mechanisms underlying the actions of MTs in OA pathogenesis. Our results showed that Mt1/Mt2 double KO causes enhanced cartilage destruction via triggering an increase in chondrocyte apoptosis. Unexpectedly, overexpression of MT2, but not MT1, induced cartilage destruction through regulatory effects on expression of matrix-degrading enzymes and cartilage ECM molecules. Our findings reveal pleiotropic roles of MTs (chondroprotection and cartilage destruction) in OA pathogenesis, and provide insights into the underlying molecular mechanisms.
Materials and methods
Human OA cartilage was sourced from individuals undergoing arthroplasty, as described previously.18 ,19 Mice were maintained under pathogen-free conditions, and all experiments involving mice were approved by the Gwangju Institute of Science and Technology Animal Care and Use Committee. Detailed experimental procedures and specific materials are described in online supplementary materials and methods and table S1, including human OA cartilage and experimental OA in mice, histology and immunohistochemistry, primary culture of articular chondrocytes and adenovirus infection, apoptosis of chondrocytes, determination of reactive oxygen species (ROS), cDNA microarray, transcription factor arrays, reporter gene assay, reverse transcription-PCR and small interfering RNA (siRNA), western blotting and statistical analysis.
Supplemental material
Results
MT1 and MT2 are upregulated in OA chondrocytes and cartilage
ZIP8 and MTF1 overexpression induced increased mRNA and protein levels of MT1 and MT2 in chondrocytes (figure 1A), consistent with our previous observations.7 MT levels were additionally elevated by other catabolic regulators of OA, including hypoxia inducible factor (HIF)-2α (encoded by Epas1),18 nicotinamide phosphoribosyltransferase (NAMPT)19 and proinflammatory cytokines, such as interleukin (IL) 1β, tumour necrosis factor-α and IL-6 (figure 1B, C; online supplementary figure S1A). MT upregulation by IL-1β was blocked upon chelation of zinc with N,N,N′,N’-Tetrakis-(2-pyridylmethyl) ethylenediamine or Ca-EDTA or knockdown of Zip8 or Mtf1 with siRNA (see online supplementary figure S1B,C). However, HIF-2α- and NAMPT-induced MT upregulation were not affected by Zip8 knockdown (see online supplementary figure S1D), indicating that these processes are independent of the zinc-ZIP8-MTF1 axis. Additionally, MT1 and MT2 protein levels were markedly increased in human OA cartilage (figure 1D) and cartilage of mouse OA caused by destabilisation of the medial meniscus (DMM) or intra-articular (IA) injection of Ad-Zip8, Ad-Mtf1, Ad-Epas1 or Ad-Nampt (figure 1E).

Upregulation of metallothioneins (MTs) in osteoarthritis (OA) chondrocytes and cartilage. (A) mRNA and protein levels of MTs in chondrocytes infected with Ad-C, Ad-Zip8 or Ad-Mtf1 (800 multiplicity of infection (MOI); n=5). (B and C) MT mRNA levels in chondrocytes infected with Ad-C (800 MOI) or the indicated MOI of Ad-Epas1 or Ad-Nampt (B; n≥5) or treated with interleukin (IL) 1β (C; n=6). (D) Alcian blue staining and immunostaining for MT1/MT2 in human OA cartilage (n=7). (E) Safranin-O and immunostaining for MT1/MT2 in cartilage of mouse OA caused by destabilisation of the medial meniscus (DMM) (8 weeks) or intra-articular injection of the indicated adenoviruses (3 weeks). Values are presented as means±SEM (*p<0.05). Scale bar: 50 μm.
Mt1/Mt2 double KO in mice potentiates DMM-induced OA via enhancement of chondrocyte apoptosis
Consistent with our previous findings,7 Mt1/Mt2 double KO mice exhibited significantly enhanced OA cartilage destruction upon DMM (figure 2A). However, we observed no substantial differences in protein levels of matrix-degrading enzymes or SOX9 between wild type (WT) and KO mice (figure 2B), indicating that potentiation of DMM-induced OA pathogenesis in KO mice is not associated with modulation of expression of matrix-degrading enzymes and cartilage ECM molecules. Similarly, IL-1β-induced upregulation of matrix-degrading enzymes and downregulation of cartilage ECM molecules were not affected in KO chondrocytes (figure 2C). We further examined the effects of deletion of Mt1/Mt2 on chondrocyte survival following exposure to mechanical stress. The percentage of apoptotic chondrocytes was significantly increased in the superficial and middle zones of articular cartilage in DMM-operated KO mice. However, this effect was not evident in the calcified region (figure 2D, E). The chondroprotective effects of MTs appear to be correlated to their capacity to regulate intracellular ROS levels. Overexpression of MT2, and to a lower degree, MT1, reduced oxidant levels in chondrocytes. Conversely, Mt1/Mt2 double KO chondrocytes exhibited a substantial increase in the basal levels of ROS and ROS levels after treatment with tert-butyl hydroperoxide, compared with WT chondrocytes (see online supplementary figure S2). These results collectively suggest that chondrocyte apoptosis and enhanced ROS levels induced by Mt1/Mt2 double KO contribute to potentiation of OA cartilage destruction.

Mt1/Mt2 double knockout (KO) in mice potentiates destabilisation of the medial meniscus (DMM)-induced osteoarthritis pathogenesis by enhancing chondrocyte apoptosis. (A) Safranin-O staining and Osteoarthritis Research Society International (OARSI) grade in DMM-operated (6 weeks) wild type (WT) and Mt1/Mt2 double KO mice (n=13). (B) Immunohistochemical analysis of sham-operated or DMM-operated WT and Mt1/Mt2 double KO mice. (C) mRNA levels of the indicated genes in WT and Mt1/Mt2 double KO chondrocytes treated with interleukin (IL) 1β (n=5). (D and E) Terminal deoxynucleotidyl transferase (TdT)-mediated deoxyuridine triphosphate (dUTP) nick end labelling (TUNEL) staining (D) and quantitation of apoptotic chondrocytes (E) in cartilage from sham-operated or DMM-operated WT and Mt1/Mt2 double KO mice (n=9). Values are presented as means±SEM (*p<0.05, **p<0.01; NS, not significant). Scale bar: 50 μm.
Overexpression of MT2, but not MT1 in joint tissues promotes OA pathogenesis in mice independently of the zinc-ZIP8-MTF1 axis
To further characterise the in vivo role of MTs in OA pathogenesis, we overexpressed MT1 or MT2 in knee joint tissues of WT mice via IA injection of Ad-Mt1 and Ad-Mt2, respectively. Consistent with our previous demonstration of effective gene delivery via IA injection of adenovirus,18–22 Ad-Mt1 and Ad-Mt2 injection led to overexpression of MTs in cartilage and other joint tissues (figure 3A, C; online supplementary figure S3A). MT1 overexpression did not induce significant changes in cartilage and synovium (figure 3B). Unexpectedly, however, cartilage destruction (figure 3D, E) and synovitis (see online supplementary figure S3B) were evident 3 weeks after IA injection of Ad-Mt2. Osteophyte formation and subchondral bone sclerosis were observed 8 weeks after Ad-Mt2 injection (figure 3E). Additional overexpression of MT1 did not alleviate MT2-induced OA pathogenesis (see online supplementary figure S3C,S3D). These results indicate that MT2, but not MT1, promotes OA pathogenesis, in contrast to the apparent chondroprotective effects of MTs in the context of Mt1/Mt2 double KO mice with DMM-induced OA pathogenesis.

Overexpression of MT2, but not MT1, in joint tissues promotes osteoarthritis (OA) pathogenesis in mice. (A and B) Representative immunostaining images for MT1/MT2 in cartilage (A) and staining and scoring of cartilage destruction and synovial inflammation (B) in wild type (WT) mice intra-articular (IA) injected with Ad-C or Ad-Mt1 (n=14). (C) Immunostaining for MT1/MT2 in cartilage of WT mice IA injected with Ad-C or Ad-Mt2 (n=12). (D and E) Safranin-O staining and scoring of OA manifestations in WT mice, 3 weeks (D) and 8 weeks (E) after IA injection of Ad-C or Ad-Mt2 (n=11). Values are presented as means±SEM (*p<0.01, **p<0.001). Scale bar: 50 μm.
Next, we examined whether MT2-induced OA pathogenesis is mediated by the zinc-ZIP8-MTF1 axis. MT2-induced OA cartilage destruction and synovitis were not affected by chondrocyte-specific cKO of Zip8 or Mtf1 in mice (see online supplementary figure S4A). Conversely, Mt1/Mt2 double KO in mice did not affect ZIP8- or MTF1-induced OA pathogenesis (see online supplementary figure S4B−F), clearly implying that MT2-induced OA pathogenesis is independent of the zinc-ZIP8-MTF1 axis.
Microarray analysis of MTs and zinc-ZIP8-MTF1 axis target genes
To elucidate the mechanisms underlying zinc-ZIP8-MTF1 axis-independent regulation of OA by MT2, we identified the target genes of MT2 and the zinc-ZIP8-MTF1 axis in chondrocytes. In response to MT2 overexpression, 385 genes were upregulated more than twofold, with 65 genes exhibiting a >50% decrease (figure 4A; online supplementary table S2). ZIP8 overexpression triggered upregulation of 213 and downregulation of 38 genes, whereas MTF1 led to upregulation of 189 genes and downregulation of 18 genes (figure 4A; online supplementary tables S3 and S4). Among the genes upregulated by MT2, 175 (45%) and 208 (54%) were also upregulated by MTF1 and ZIP8, respectively (figure 4A; online supplementary tables S3 and S4). In total, 120 genes were upregulated in common by MT2, ZIP8 and MTF1 (figure 4A; online supplementary table S5). Our microarray analyses collectively revealed that almost half of the MT2 target genes are independent of the zinc-ZIP8-MTF1 axis. Considering that the vast majority of genes jointly regulated by ZIP8, MTF1 and MT2 include general OA-associated markers, the MT2-specific targets suggest zinc-ZIP8-MTF1 axis-independent regulation of OA pathogenesis by MT2 (figure 4B).
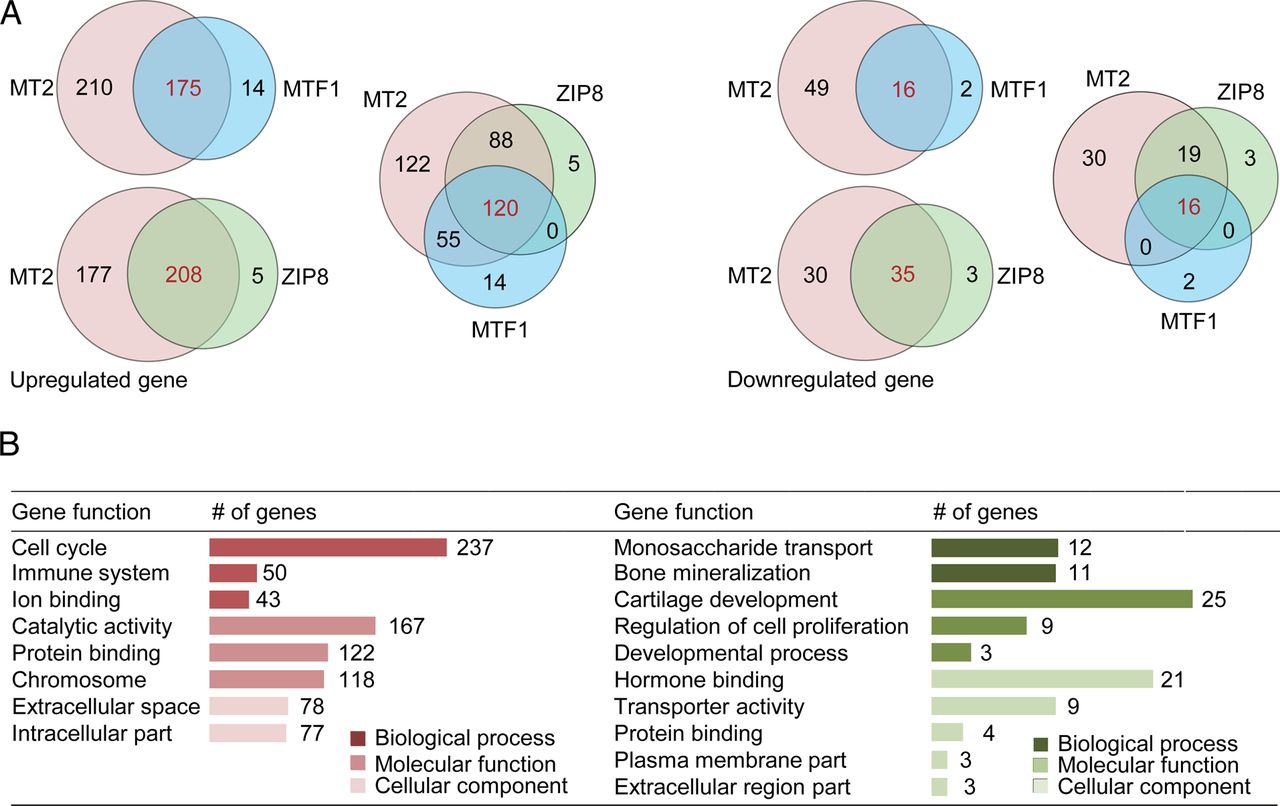
Microarray analyses of MT2, MTF1 and ZIP8 target genes. (A) Venn diagram summarising the responsive genes that are differentially upregulated (left; p<0.05; fold change ≥2) or downregulated (right; p<0.05; fold change ≤0.5) upon MT2, MTF1 or ZIP8 overexpression in chondrocytes. (B) Functional categories of MT2 target genes.
Genetic deletion of Mt1/Mt2 did not significantly alter the gene expression profile in chondrocytes. Only 5 genes exhibited a >1.5-fold increase while 29 genes showed a >30% decrease (see online supplementary table S6). Meanwhile, in response to MT1 overexpression, only 14 genes were upregulated >1.5-fold and 34 genes exhibited a >30% decrease (see online supplementary tables S7). However, among the genes differentially regulated by MT1, no genes were jointly upregulated and downregulated by MT2, further supporting a distinct role of MT2 (see online supplementary table S7).
MT2 upregulates matrix-degrading enzymes and downregulates cartilage matrix molecules
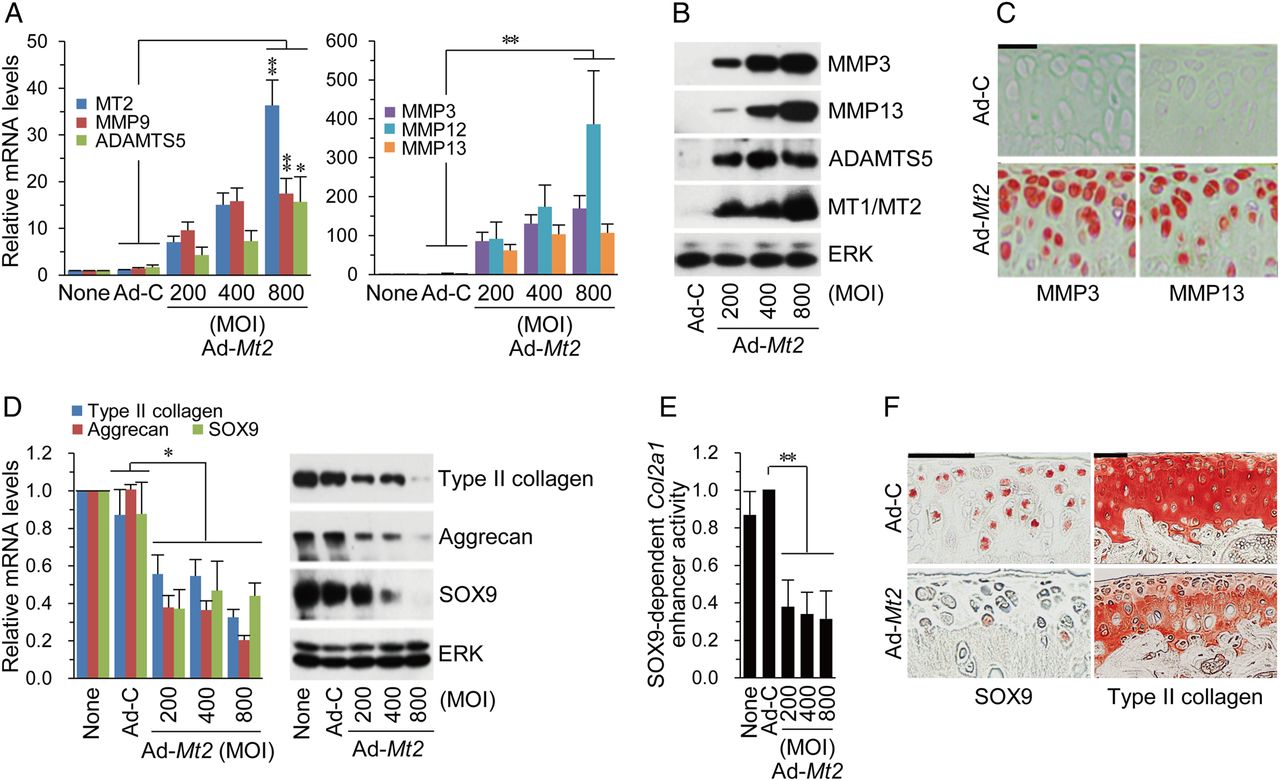
To elucidate the regulatory mechanisms underlying MT2-induced OA, we evaluated the effects of MT2 on the expression of matrix-degrading enzymes and cartilage ECM molecules. MT2 overexpression in chondrocytes led to increased mRNA and protein levels of matrix-degrading enzymes, including MMP3, MMP13 and ADAMTS5 (figure 5A, B). Consistently, IA injection of Ad-Mt2 caused a marked increase in MMP3 and MMP13 in cartilage tissue (figure 5C). In addition, MT2 overexpression induced a significant reduction in SOX9, type II collagen, and aggrecan mRNA and protein levels (figure 5D) in association with a decrease in SOX9-dependent Col2a1 enhancer activity (figure 5E). Consistently, SOX9 and type II collagen protein levels in cartilage were significantly reduced following Ad-Mt2 IA injection (figure 5F).

MT2 modulates the expression of matrix-degrading enzymes and cartilage matrix molecules in chondrocytes. (A and B) mRNA (A) and protein (B) levels of matrix-degrading enzymes in chondrocytes infected with Ad-C (800 multiplicity of infection (MOI)) or the indicated MOI of Ad-Mt2 (n≥4). (C) MMP3 and MMP13 immunostaining in cartilage tissues of wild type (WT) mice intra-articular (IA) injected with Ad-C or Ad-Mt2 (n=12). (D and E) mRNA and protein levels of extracellular matrix molecules and SOX9 (D) and SOX9-dependent Col2a1 enhancer activity (E) in chondrocytes infected with Ad-C or Ad-Mt2 (n≥4). (F) SOX9 and type II collagen immunostaining in cartilage of WT mice IA injected with Ad-C or Ad-Mt2. Values are presented as means±SEM (*p<0.05, **p<0.001). Scale bar: 50 μm.
In contrast to MT2, overexpression of MT1 in chondrocytes did not markedly alter the expression patterns of matrix-degrading enzymes or ECM molecules (see online supplementary figure S5A). Furthermore, neither overexpression nor siRNA-mediated knockdown of MT1 affected MT2-mediated regulation of these genes (see online supplementary figure S5B–D). Knockdown of Zip8 or Mtf1 did not affect MT2-induced pathogenic gene expression (see online supplementary figure S6A,B), supporting the zinc-ZIP8-MTF1 axis-independent action of MT2. We previously identified MMP3, MMP9, MMP12 and MMP13 as target genes of HIF-2α,18 and MMP3, MMP12 and MMP13 as targets of NAMPT.19 Since transcription of these genes and HIF-2α was induced upon MT2 overexpression, we further examined whether HIF-2α and NAMPT are involved in MT2-mediated pathogenic gene expression. Notably, knockdown of HIF-2α or NAMPT did not influence MT2-induced upregulation of matrix-degrading enzymes or downregulation of ECM molecules in chondrocytes (see online supplementary figure S6C–E).
NF-κB and AP-1 mediate MT2-induced OA pathogenesis
Next, to elucidate the mechanisms underlying MT2-mediated modulation of matrix-degrading enzymes and ECM molecules, we identified the transcription factors modulated by MT2 in chondrocytes. Among the transcription factors examined, STAT1/2, ATF2, nuclear factor-κ B (NF-κB), RBP-JK, IRF1 and activator protein-1 (AP-1) showed >twofold increase in transcriptional activity following Ad-Mt2 infection (figure 6A). Neither pharmacological inhibition of the Notch pathway (RBP-JK) with N-(N-(3,5-difluorophenacetyl)-l-alanyl)-S-phenylglycine t-butyl ester nor knockdown of STAT1, STAT2, ATF2 or IRF1 with specific siRNAs affected MT2-induced expression of matrix-degrading enzymes or ECM molecules (see online supplementary figure S7A). However, inhibition of NF-κB with SC-51423 led to significant ablation of MT2-induced upregulation of matrix-degrading enzymes (figure 6B). AP-1 inhibition with Tanshinone IIA (T-IIA)24 additionally blocked upregulation of MMP9 and MMP13, with no apparent effects on MMP3, MMP12 or ADAMTS5 (figure 6B). Downregulation of cartilage matrix molecules was reversed upon inhibition of NF-κB, but not AP-1 (figure 6C). Furthermore, IA co-injection of SC-514 or T-IIA significantly blocked MT2-induced cartilage destruction (figure 6D) and synovitis (see online supplementary figure S7B). These results suggest that NF-κB and AP-1 mediate MT2-induced OA pathogenesis.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Nuclear factor-kappa B (NF-κB) and activator protein-1 (AP-1) mediate MT2-induced osteoarthritis (OA) pathogenesis. (A) Transcriptional activity of the indicated transcription factors in chondrocytes infected with Ad-C or Ad-Mt2 (800 multiplicity of infection (MOI); n=2). (B and C) mRNA levels of the indicated genes in chondrocytes infected with Ad-C or Ad-Mt2 (800 MOI) in the absence or presence of SC-514 or Tanshinone IIA (T-IIA) (n=6). (D) Safranin-O staining and Osteoarthritis Research Society International (OARSI) grade in mice intra-articular injected with Ad-C or Ad-Mt2 with or without SC-514 or T-IIA (n=12). Values are presented as means±SEM (*p<0.05, **p<0.01, ***p<0.001; NS, not significant). Scale bar: 50 μm.
Discussion
The hallmark of OA is an imbalance between chondrocyte catabolism and anabolism. Biochemical pathway activation in chondrocytes leads to cartilage destruction by MMPs and ADAMTS.25–27 During OA pathogenesis, cartilage proteoglycan is depleted through cessation of ECM synthesis. Increased chondrocyte apoptosis also occurs in cartilage in response to a variety of OA factors, including mechanical stress and proinflammatory cytokines, which results in a decrease in cellularity and is associated with loss of ability to maintain cartilage homoeostasis. In fact, the extent of chondrocyte apoptosis is correlated with severity of OA.6 ,21 ,28
MTs play diverse roles in metal detoxification, antioxidation, regulation of Zn2+-dependent proteins, immune defence and cell differentiation.29 ,30 Notably, MTs regulate cellular Zn2+ levels, and impaired Zn2+ homoeostasis is implicated in the pathophysiology of aging and various diseases.31 ,32 Our current findings provide new insights into the underlying mechanisms of regulation of OA pathogenesis by MTs, demonstrating catabolic and anabolic functions as well as antiapoptotic actions of MTs. We identified HIF-2α, NAMPT and proinflammatory cytokine pathways as well as the zinc-ZIP8-MTF1 axis as upstream regulators of MTs and showed their contribution to increased expression of MTs in OA cartilage. Data from our loss-of-function study using Mt1/Mt2 double KO mice suggest that MTs are necessary for maintaining cartilage homoeostasis through suppression of apoptosis and oxidative stress. OA chondrocytes are characterised by increased Zn2+ content,7 and intracellular zinc influx is associated with apoptosis of growth plate chondrocytes.33 Therefore, upregulation of MT proteins may be essential for alleviating apoptotic stress induced by accelerated zinc flux during OA pathogenesis. Significant accumulation of oxidative stress has been reported in aging and OA cartilage.34 Our results indicate that MTs modulate the intracellular ROS level, supporting the conclusion that MTs exert protective roles against apoptosis by scavenging and controlling cellular oxidative stress.
Unexpectedly, we found that chronic, aberrant upregulation of MT2 contributes to OA pathogenesis. MT2 serves as a critical regulator of OA by upregulating matrix-degrading enzymes and downregulating cartilage-specific ECM proteins. Additionally, MT2 activates a variety of transcription factors, and NF-κB and AP-1 are involved in the transcriptional regulation of these OA-associated genes. A previous study implicated MT2 activation of NF-κB and AP-1 in transcription of MMP9 in breast cancer cells.35 Moreover, a role for MTs in transcriptional activity of NF-κB has been demonstrated in mouse fibroblasts.36 Together with previous studies, our current results highlight the specific role of MT2 as a modulator of transcription factor activity. Differential metal binding affinities of MT1 and MT2 towards divalent metal ions may account for their distinct physiological functions. For instance, MT2 plays a primary role in maintaining the homoeostasis of zinc and copper ions while MT1 has a detoxification function via its superior affinity for toxic heavy metals, such as cadmium.37
In association with their detoxification and antioxidant roles, therapeutic utility of MTs for inflammatory diseases, such as rheumatoid arthritis, has been suggested.38 Consistent with earlier findings, our results disclosed antiapoptotic roles of MTs in chondrocytes, supporting their therapeutic potential. However, chronic increase in the MT2 level unexpectedly promoted OA pathogenesis by causing an imbalance in homoeostasis between matrix catabolism and anabolism. These variable effects of MTs provide a caveat for their therapeutic consideration in OA.
References
Footnotes
Handling editor Tore K Kvien
Contributors YW: Study design, data acquisition, data analysis and interpretation, manuscript preparation and approval; YS, C-HC, YC, and C-WH: data acquisition, analysis and interpretation, and manuscript approval; J-HK and J-SC: funding acquisition, study design, data interpretation, manuscript preparation and approval. J-HK and J-SC take responsibility for the integrity of this work.
Funding This work was supported by grants from the National Research Foundation of Korea (2007-0056157 and 2013R1A2A1A01009713), the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (H114C3484), and the Silver Health Bio Research Center of Gwangju Institute of Science and Technology to J-SC. J-HK was supported by grants from the National Research Foundation of Korea (2015M3A9E6028674) and the National R&D Program for Cancer Control (1520070) funded by the Ministry of Health & Welfare. YW was supported by a Global PhD Fellowship Program through the National Research Foundation of Korea funded by the Ministry of Education (2014H1A2A1017835).
Competing interests None declared.
Patient consent Obtained.
Ethics approval The Institutional Review Board of the Wonkwang University Hospital approved the use of human materials. All animal experiments were approved by the Gwangju Institute of Science and Technology Animal Care and Use Committee.
Provenance and peer review Not commissioned; externally peer reviewed.