Article Text

Abstract
Objectives This study compared the efficacy and safety of subcutaneous (SC) versus intravenous (IV) formulations of tocilizumab in patients with rheumatoid arthritis with an inadequate response to disease-modifying antirheumatic drugs (DMARD).
Methods Patients (n=1262) were randomly assigned to receive tocilizumab-SC 162 mg weekly+placebo-IV every 4 weeks or tocilizumab-IV 8 mg/kg every 4 weeks+placebo-SC weekly in combination with traditional DMARD. The primary outcome was to demonstrate the non-inferiority of tocilizumab-SC to tocilizumab-IV with regard to the proportion of patients in each group achieving an American College of Rheumatology (ACR) 20 response at week 24 using a 12% non-inferiority margin (NIM). Secondary outcomes were disease activity score using 28 joints (DAS28), ACR responses, health assessment questionnaire scores and safety assessments.
Results At week 24, 69.4% (95% CI 65.5 to 73.2) of tocilizumab-SC-treated patients versus 73.4% (95% CI 69.6 to 77.1) of tocilizumab-IV-treated patients achieved an ACR20 response (weighted difference between groups −4.0%, 95% CI −9.2 to 1.2); the 12% NIM was met. ACR50/70 responses, DAS28 and physical function improvements were comparable between the tocilizumab-SC and tocilizumab-IV groups. The safety profiles of tocilizumab-SC and tocilizumab-IV were similar, and the most common adverse event was infection. Injection-site reactions (ISR) occurred more frequently in the tocilizumab-SC group than in the tocilizumab-IV (placebo-SC) group. No anaphylaxis was reported over the 24 weeks.
Conclusions Tocilizumab-SC 162 mg weekly demonstrated comparable efficacy to tocilizumab-IV 8 mg/kg. The safety profile of tocilizumab-SC is consistent with the known and well-established safety profile of tocilizumab-IV, with the exception of a higher incidence of ISR, which were more common with tocilizumab-SC administration.
- Rheumatoid Arthritis
- DMARDs (biologic)
- Disease Activity
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) is a chronic, progressive, systemic autoimmune disease associated with joint inflammation. Advances in RA treatments have been made through the introduction of biological therapies, including anti-tumour necrosis factor (TNF) inhibitors, interleukin (IL)-1 receptor and IL-6 receptor (IL-6R) antagonists, an anti-CD20 agent and a T-cell co-stimulation modulator.1 Although these treatment options reduce disease activity, none are effective in all patients. While a patient's disease status and overall health should be central when choosing a therapy, differences in the route of administration and safety profiles of the agent can also affect the probability of a favourable response.1
Tocilizumab is a recombinant humanised anti-IL-6R monoclonal antibody that blocks IL-6 from binding to the soluble and membrane-bound IL-6R and was initially developed as an intravenous (IV) infusion. The efficacy and safety of tocilizumab-IV were previously demonstrated as monotherapy and in combination with disease-modifying antirheumatic drugs (DMARD) in adult patients with RA in five phase 3 clinical trials.2 Tocilizumab is currently approved as an IV formulation for the treatment of RA, including in the USA and Europe.
A subcutaneous (SC) formulation of tocilizumab would offer patients an additional option that may allow self-administration. The tocilizumab-SC dose was selected based on pharmacokinetic/pharmacodynamic and limited efficacy and safety data from phase 1/2 studies (see supplementary figure S1, available online only).3 To characterise further the efficacy and safety of tocilizumab-SC, the SUMMACTA study compared tocilizumab-SC 162 mg weekly versus tocilizumab-IV 8 mg/kg every 4 weeks in adult patients with RA who have had an inadequate response to one or more DMARD.
Patients and methods
Participants
Patients (≥18 years of age) with RA (≥6 months, revised 1987 American College of Rheumatology (ACR) criteria) who met the following major criteria were included: swollen joint count of 4 or greater (66-joint count) and tender joint count of 4 or greater (68-joint count) at screening and baseline, C-reactive protein (CRP) 10 mg/L or greater and/or erythrocyte sedimentation rate (ESR) 28 mm/h or greater at screening. Patients must have received one or more traditional DMARD at a stable dose for 8 weeks or longer before baseline. Patients were required to have had an inadequate response to DMARD (up to 20% of patients may have failed one or more anti-TNF). Before random assignment, patients discontinued all biological DMARD, including etanercept for 2 weeks or longer and infliximab, certolizumab, golimumab or adalimumab for 8 weeks or longer. Concomitant oral glucocorticoids (≤10 mg/day prednisone or equivalent) and non-steroidal anti-inflammatory drugs (up to the maximum recommended dose) were permitted if patients were on a stable dose for 4 weeks or longer before baseline.
Major exclusion criteria included ongoing rheumatic or inflammatory joint diseases other than RA, any active infections, history of malignancy, positive hepatitis B surface antigen or hepatitis C antibody, serious allergies to biological agents, previous treatment with tocilizumab, alkylating agents or cell-depleting therapies or treatment with any investigational agent at less than 4 weeks of screening, and intra-articular or parenteral glucocorticoids or immunisation with a live/attenuated vaccine less than 4 weeks before baseline. Tuberculosis screening was managed according to local practice.
Study design
SUMMACTA was a 2-year, randomised, double-dummy, active-controlled, parallel-group, phase 3 multicentre trial with a double-blind period of 24 weeks followed by an open-label period of 72 weeks (results presented separately). Patients were stratified by geographical region and body weight category (<60, ≥60 to <100 or ≥100 kg). During the double-blind period, patients were randomly assigned 1 : 1 to receive 162 mg of tocilizumab-SC 162 mg per week+placebo-IV every 4 weeks or tocilizumab-IV 8 mg/kg every 4 weeks+placebo-SC weekly for 24 weeks (see supplement, available online only). Tocilizumab-SC/placebo injections were administered by prefilled syringe. After the first four treatments, subcutaneous injections could be administered at home by patients or caregivers; home injection information was recorded on diary cards. Dose modification to subcutaneously every 2 weeks or intravenously 4 mg/kg every 4 weeks or dose interruption was permitted for safety concerns.
Outcomes and assessments
The primary outcome was to evaluate the non-inferiority of tocilizumab-SC 162 mg weekly to tocilizumab-IV 8 mg/kg regarding the proportion of patients in each group who achieved an ACR20 response at week 24. Secondary endpoints included the proportion of patients who achieved an ACR50/70 response, remission based on the disease activity score using 28 joints (DAS28 <2.6) and a decrease from baseline of 0.3 or greater in the health assessment questionnaire–disability index (HAQ-DI) at week 24. Subgroup analyses for ACR responses and DAS28 remission were conducted across the three weight categories. Due to the non-inferiority design, the per-protocol (PP) population was used for the primary, secondary and subgroup analyses. The PP population was a subset of the intent-to-treat (ITT) population and excluded patients with major protocol violations that could potentially affect efficacy outcomes. All patients signed informed consent documents, and the study was conducted in accordance with the Declaration of Helsinki and good clinical practice.
Safety
The safety population included patients who received one or more dose of tocilizumab and had one or more post-dose safety assessment. Safety assessments included adverse events (AE), laboratory assessments, physical examination and vital signs. Laboratory monitoring was conducted at weeks 2 and 4 and every 4 weeks thereafter.
Pharmacokinetics and pharmacodynamics
Patients who provided one or more evaluable pharmacokinetic sample were included in the pharmacokinetic population. In the main study, the observed pre-dose tocilizumab concentration (Ctrough) over 24 weeks was assessed; in the pharmacokinetic substudy at week 20, the mean area under the curve (AUC), maximum plasma concentration (Cmax) and Ctrough. pharmacodynamic parameters included CRP and ESR, were assessed as analysed in the safety population.
Immunogenicity
Blood samples were taken at baseline and throughout the study for anti-drug antibody assessment. Positive samples from the initial screening assay were analysed by a confirmation assay for specificity. The assays were performed as previously described using a bridging ELISA.4 If the confirmation assay was positive, an inhibition ELISA was performed to evaluate the neutralising potential of the anti-tocilizumab antibody.
Statistical methods
The time point for the primary analysis of the ACR20 response was week 24. To claim non-inferiority, the lower bound of the 95% CI for the difference in ACR responses (tocilizumab-SC 162 mg weekly minus tocilizumab-IV 8 mg/kg) had to be greater than −12%. A non-inferiority margin of 12% was defined based on the results observed in the tocilizumab-IV trials.5–7
For the primary analysis, the 95% CI of the weighted difference in ACR responses between the groups at week 24 was calculated using Cochran–Mantel–Haenszel analysis. This difference was adjusted for the stratification factors of region and body weight. For all secondary endpoints, the differences in treatment effects and 95% CI were reported. Sample size determination is outlined in the supplement (available online only).
Results
Patient disposition and baseline characteristics
Of the 1262 patients randomly assigned, 631 received tocilizumab-SC 162 mg weekly+placebo-IV and 631 received tocilizumab-IV 8 mg/kg+placebo-SC (figure 1). The PP population comprised 1095 patients (n=558, tocilizumab-SC 162 mg weekly; n=537, tocilizumab-IV 8 mg/kg). The most common protocol violation leading to PP exclusion was a non-stable dose of DMARD for both groups.

Patient disposition over 24 weeks. TCZ-IV, intravenous tocilizumab; TCZ-SC, subcutaneous tocilizumab. AE, adverse events; qw, weekly.
Patient baseline demographics and clinical characteristics were balanced across the tocilizumab-SC and tocilizumab-IV groups in the PP (table 1) and safety populations (data not shown). The mean RA duration, tender joint counts, swollen joint counts and DAS28 scores were comparable between groups.
Baseline demographics (per-protocol population)
The prevalence of previous and current medications for RA was similar between groups. The proportion of patients who had inadequate responses to anti-TNF inhibitors was 22.5% in the tocilizumab-SC group and 21.6% in the tocilizumab-IV group; 79.7% of the tocilizumab-SC group and 81.5% of the tocilizumab-IV group received methotrexate.
Efficacy
The study met its primary endpoint by demonstrating the non-inferiority of tocilizumab-SC 162 mg weekly to tocilizumab-IV 8 mg/kg. The proportion of tocilizumab-SC patients achieving an ACR20 response at week 24 was 69.4% (95% CI 65.5 to 73.2); for tocilizumab-IV patients, it was 73.4% (95% CI 69.6 to 77.1; figure 2A). The difference between groups was −4.0% (95% CI −9.2 to 1.2), confirming the non-inferiority of tocilizumab-SC to tocilizumab-IV. The robustness of the primary endpoint analysis was supported by analysis in the ITT population: −2.7% (95% CI −7.6 to 2.2; see supplementary figure S2, available online only).
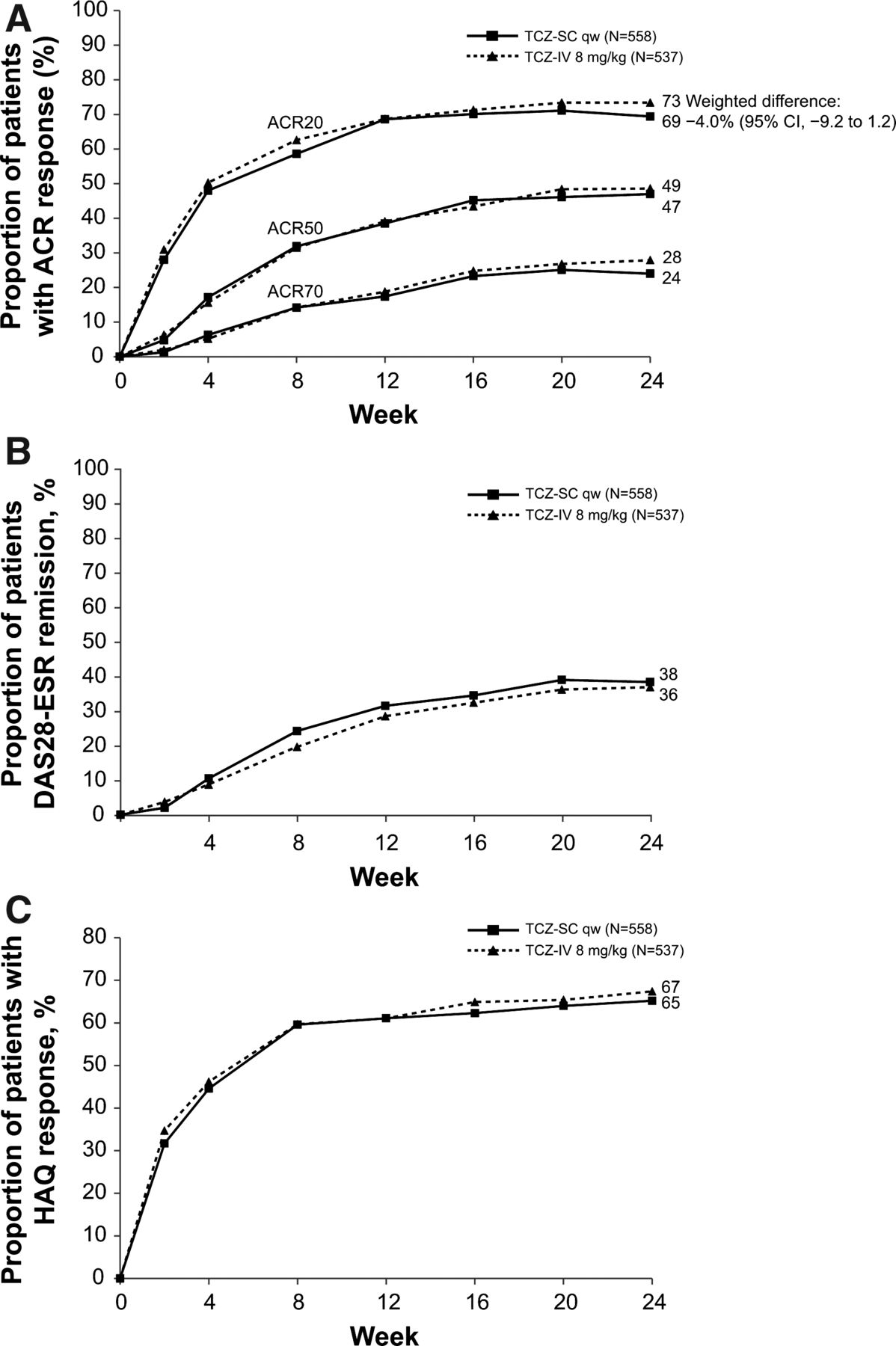
Disease activity and physical function over 24 weeks for patients in the per-protocol (PP) population. (A) Proportion of patients in the PP population treated with either subcutaneous tocilizumab (TCZ-SC; n=558) or intravenous tocilizumab (TCZ-IV; n=537) achieving 20%, 50% and 70% improvements per American College of Rheumatology criteria (ACR20, ACR50 and ACR70) over 24 weeks. (B) Proportion of patients achieving remission based on disease activity score using 28 joints (DAS28) based on erythrocyte sedimentation rate (ESR <2.6) over 24 weeks. (C) Proportion of patients achieving a health assessment questionnaire (HAQ) response (improvement of ≥0.3 from baseline) over 24 weeks. qw, weekly.
ACR20, ACR50 and ACR70 response rates over 24 weeks were similar between groups (figure 2A). The weighted differences in the proportion of ACR50 and ACR70 responders at week 24 were −1.8% (95% CI −7.5 to 4.0) and −3.8% (95% CI −9.0 to 1.3), respectively.
The proportion of patients who achieved DAS28 remission over 24 weeks was comparable between groups (figure 2B). The weighted difference in the proportion of patients achieving DAS28 remission at week 24 was 0.9% (95% CI −5.0 to 6.8). In post-hoc analyses, the proportion of patients who achieved simple disease activity index, clinical disease activity index and Boolean remission were also similar between groups (see supplementary figure S3, available online only).
The proportion of patients who achieved a decrease of 0.3 or greater in HAQ-DI score from baseline was similar between groups (figure 2C). The weighted difference in the proportion of patients achieving a decrease of 0.3 or greater in HAQ-DI score from baseline was −2.3% (95% CI −8.1 to 3.4).
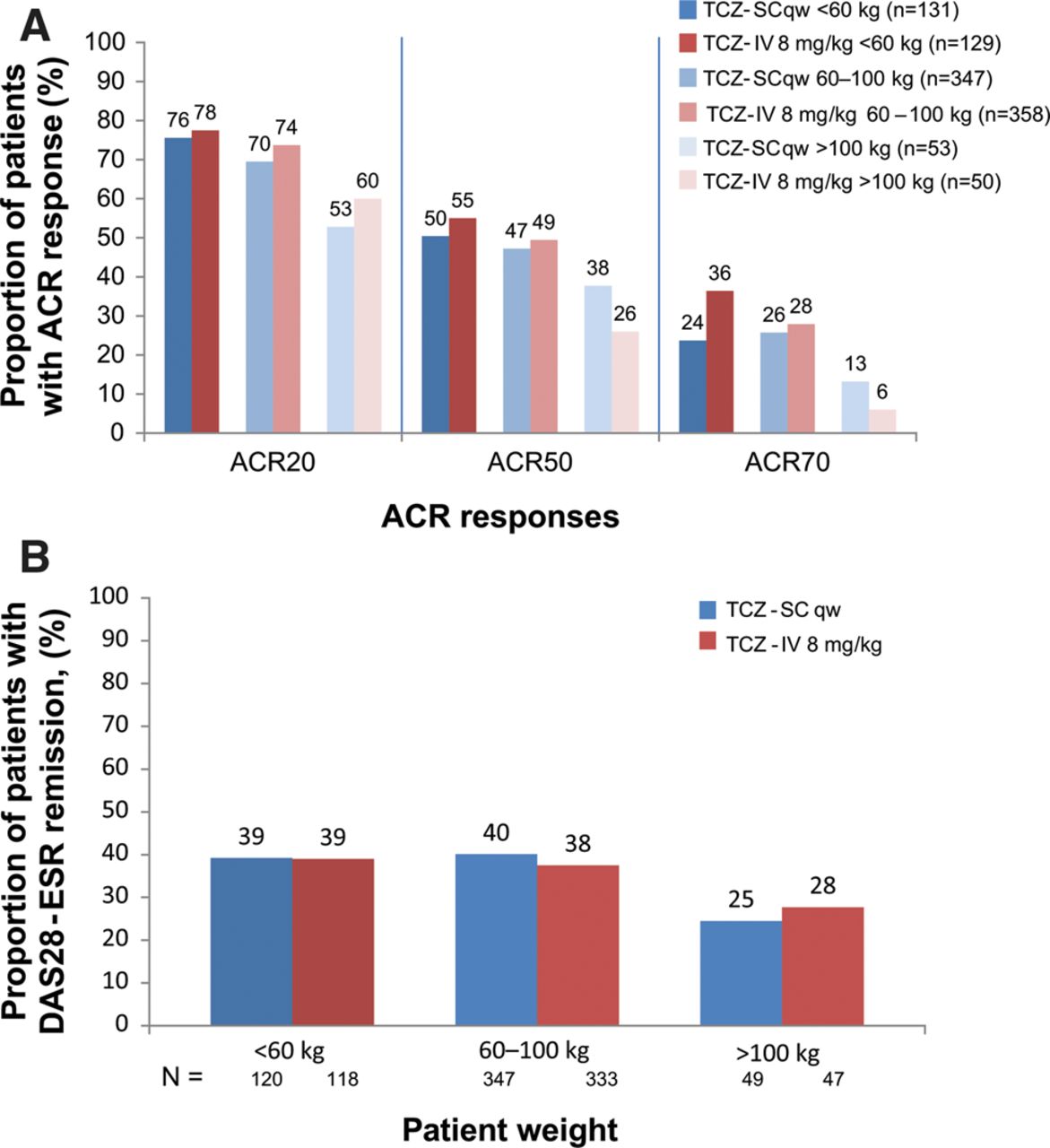
The proportion of patients achieving an ACR20/50/70 response (figure 3A) or DAS remission (figure 3B) was similar between the groups across the three body weights. The response in the heaviest weight category (≥100 kg) of both arms was lower and had more variation, which may be due to the lower number of patients in this weight category.
{kind=link}
{kind=link}
{kind=link}
Proportion of patients stratified by weight in the per-protocol population treated with either subcutaneous tocilizumab (TCZ-SC; n=558) or intravenous tocilizumab (TCZ-IV; n=537) (A) achieving 20%, 50% and 70% improvements per American College of Rheumatology criteria (ACR20, ACR50 and ACR70) over 24 weeks and (B) achieving remission based on disease activity score using 28 joints (DAS28) based on erythrocyte sedimentation rate (ESR <2.6) over 24 weeks. qw, weekly.
Pharmacokinetics and pharmacodynamics
The pharmacokinetic profiles of tocilizumab following subcutaneous and intravenous dosing varied due to the different routes of administration. Serum trough tocilizumab levels increased more rapidly in the tocilizumab-SC group than in the tocilizumab-IV group (see supplementary figure S4, available online only). With subsequent dosing, the trough tocilizumab levels plateaued by week 12 in both groups. The observed steady-state Ctrough (±SD) at week 24 was 42 (±27.4) μg/mL following tocilizumab-SC dosing and 18 (±14.2) μg/mL following tocilizumab-IV dosing.
Despite higher trough tocilizumab levels for tocilizumab-SC, the mean AUC and Cmax of tocilizumab at steady state were higher for tocilizumab-IV (see supplementary figure S5, available online only). Thirty-four patients participated in a pharmacokinetic substudy with more frequent sampling at week 20; 26 patients had sufficient data for analysis (tocilizumab-SC, n=13; tocilizumab-IV, n=13) (see supplementary figure S5, available online only). At steady state from weeks 20 to 24, the mean AUC20–24wk was 30 168 μg h/mL for tocilizumab-SC and 41 304 μg h/mL for tocilizumab-IV. The mean Cmax values (±SD) at week 20 were 52.7 (±27.3) μg/mL for tocilizumab-SC and 233 (±117) μg/mL for tocilizumab-IV.
CRP levels decreased in both groups after the first dose of tocilizumab (see supplementary figure S6, available online only). Thereafter, in both groups, CRP remained below the upper limit of normal (ULN; 0.99 mg/dL) to week 24. The time course of CRP for tocilizumab-SC was comparable to that for tocilizumab-IV, although there was a trend towards slightly lower CRP levels in the tocilizumab-SC group. Similar results were observed for ESR (see supplementary figure S7, available online only).
Safety
The safety profile was similar between groups (safety population), except for more injection-site reactions (ISR) in the tocilizumab-SC group (table 2). Rates of AE, serious adverse events (SAE) and discontinuation due to AE were similar between groups. The proportion of patients in each group with a dose modification or interruption due to an AE was comparable (see supplementary table S1, available online only). The frequency of infection was 36.0% in the tocilizumab-SC group and 39.1% in the tocilizumab-IV group. The most common AE was infection; among them, the most commonly reported preferred term was upper respiratory tract infection (7.3% tocilizumab-SC and 11.6% tocilizumab-IV). The most common SAE was infection (1.4% in both groups). Serious infections of pneumonia occurred in two patients in each group and bacterial arthritis occurred in two patients in the tocilizumab-IV group. The most common reason for withdrawal due to AE in both groups was infection (1.1% tocilizumab-SC and 1.3% tocilizumab-IV). No obvious differences were observed between treatment groups for incidences of AE and SAE across the three weight categories (see supplementary table S2, available online only).
Safety summary (safety population)
No deaths were reported for patients receiving tocilizumab-SC. One death was reported in the tocilizumab-IV group, attributable to sepsis due to bacterial arthritis.
Three SAE of malignancy (<1%) occurred in the tocilizumab-SC group and one (<1%) in the tocilizumab-IV group. In the tocilizumab-SC group, breast cancer was diagnosed in two patients and a brain neoplasm in one patient. In the tocilizumab-IV group, squamous cell carcinoma was diagnosed in one patient.
ISR were more common in the tocilizumab-SC group (10.1%) than in the tocilizumab-IV group (2.4%), in which patients also received placebo-SC. All ISR were non-serious and common terminology criteria for adverse events grade 1 or 2; none required dose interruption or withdrawal.
No anaphylaxis events were reported. Five SAE were observed within 24 h of infusion or injection and evaluated as ‘related’ to study treatment; of these, three were medically consistent with hypersensitivity and led to early study withdrawal (two events in the tocilizumab-SC group and one event in the tocilizumab-IV group). All events resolved without sequelae.
After initiation of tocilizumab, the majority of shifts in ALT and AST from normal at baseline were threefold or less the ULN; ALT shifts occurred in more patients in the tocilizumab-SC group compared with the tocilizumab-IV group (46% vs 39%, see supplementary table S3, available online only). No differences between groups were observed for ALT and AST shifts from normal at baseline to a value between more than three times and five times or less ULN, or from normal to more than five times ULN. One patient in the tocilizumab-SC group and three patients in the tocilizumab-IV group had sustained consecutive ALT elevations (an elevation from the time of the first elevation to the last record). One patient in the tocilizumab-SC group and two patients in the tocilizumab-IV group had sustained consecutive AST elevations. Five patients (0.8%) in the tocilizumab-SC group and seven patients (1.1%) in the tocilizumab-IV group prematurely discontinued because of elevated liver transaminases. Among patients with a low neutrophil count after initiating treatment, most experienced common terminology criteria grade 1 or 2 neutropenia (see supplementary table S3, available online only). Grades 1 and 2 events of neutropenia were reported for a slightly higher proportion of patients in the tocilizumab-SC group compared with the tocilizumab-IV group. No differences were observed between groups for grades 3 or 4 neutropenia. One patient in the tocilizumab-SC group experienced a sustained consecutive neutrophil decline. For patients with a low platelet count, almost all events were grade 1. The proportion of patients with an increase in total cholesterol from less than 200 mg/dL at baseline to more than 200 mg/dL at the last observation was slightly higher in the tocilizumab-SC group than in the tocilizumab-IV group (50% vs 42%). The largest categorical shift in total cholesterol (from <200 to ≥240 mg/dL) was more common in tocilizumab-SC patients than in tocilizumab-IV patients (7% vs 4%). Clinically relevant shifts in low-density lipoprotein cholesterol, high-density lipoprotein cholesterol and triglyceride levels were similar between groups.
Immunogenicity
Five patients in each group were positive (0.8% and 0.8%) for both the confirmation and neutralising assays post-baseline. No patients with serious hypersensitivity events developed anti-tocilizumab antibodies. One patient in the tocilizumab-SC group with anti-tocilizumab antibodies had an ISR. No patients with detected anti-tocilizumab antibodies withdrew because of an insufficient therapeutic response or loss of efficacy (defined as patients from the ITT population who withdrew due to insufficient therapeutic response or who experienced an ACR50 or DAS–ESR-based European League Against Rheumatism) good response before withdrawal). Based on the limited number of patients who developed anti-tocilizumab antibodies, no impact of antibodies on the pharmacokinetics of tocilizumab was observed.
Discussion
Tocilizumab-IV is an efficacious therapy with an acceptable risk/benefit profile for patients with RA. The SUMMACTA study analysed whether tocilizumab-SC 162 mg weekly was non-inferior to tocilizumab-IV 8 mg/kg in patients with RA with an inadequate response to DMARD, which may have included one or more anti-TNF inhibitors. The primary endpoint was met by demonstrating that the ACR20 response at week 24 for patients treated with tocilizumab-SC was non-inferior to that of patients treated with tocilizumab-IV. The safety profiles of tocilizumab-SC and tocilizumab-IV were similar, except for a higher incidence of ISR more commonly observed with tocilizumab-SC administration.
The ACR20 response rate for the tocilizumab-IV patients in the PP population was higher than that observed in previous tocilizumab-IV studies.5–8 Baseline disease characteristics were comparable with previous tocilizumab-IV studies, so the higher ACR20 response rate in this study may reflect that all patients received active tocilizumab treatment, whereas previous tocilizumab-IV studies included a placebo group or other comparator during the double-blind phase.
The CRP time course was comparable between groups. There was a trend towards slightly lower CRP levels in the tocilizumab-SC group, which was consistent with the higher Ctrough concentrations in the tocilizumab-SC group over 24 weeks. These results were similar to those of previous phase 1/2 studies for tocilizumab-SC 162 mg weekly, thus validating the assumptions made for the dose assessed.
The safety profiles between groups were similar in this study and consistent with previous studies of long-term administration of tocilizumab-IV, with infections being the most common AE and SAE;5–8 consistent with other subcutaneous RA treatments, higher ISR rates were reported in tocilizumab-SC than in tocilizumab-IV. The incidence rate (10.1%) of ISR in the tocilizumab-SC group was similar to that reported at 24 weeks in other studies in patients with RA who received subcutaneous anti-TNF inhibitors.9 ,10
Comparison of the incidence of antibodies to tocilizumab-SC with other subcutaneous RA therapies is difficult because the measurement of antibody positivity (including neutralising antibodies) is highly dependent on assay sensitivity, specificity and methodology. In this study, the proportion of patients in both groups who developed anti-tocilizumab antibodies was low and comparable; no correlation was observed between antibody development and AE or clinical response. Therefore, the immunogenicity potential following tocilizumab-SC treatment was considered low based on available data.
A limitation of the data is that long-term efficacy and safety have not been explored. Longer observation from this study and additional data from other studies with tocilizumab-SC will provide further information related to immunogenicity and AE.
In summary, the non-inferiority of tocilizumab-SC 162 mg weekly to tocilizumab-IV 8 mg/kg every 4 weeks was demonstrated. Tocilizumab-SC demonstrated efficacy and clinical safety profiles comparable with those of tocilizumab-IV, with the exception of a higher incidence of ISR more commonly seen with tocilizumab-SC. The tocilizumab-SC formulation could provide an additional, more convenient administration option and opportunity for home injection for patients with RA.
Acknowledgments
The authors wish to thank all investigators and patients who participated in the study and all members of the WA22762 study team. Support for third-party writing assistance for this manuscript, furnished by Denise Kenski, PhD of Health Interactions, was provided by F. Hoffmann-La Roche, Ltd.
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
-
Correction notice This article has been corrected since it was published Online First. Figure 2A has been amended, and the following sentence amended to read: The most common AE was infection; among them, the most commonly reported preferred term was upper respiratory tract infection (7.3% tocilizumab-SC and 11.6% tocilizumab-IV). The most common SAE was infection (1.4% in both groups).
-
Contributors LR and MB designed the study, analysed and interpreted the data. PL analysed and interpreted the data. GRB, AR-R, AC, SH, PL, DF, MJR, GR, CL and EFM were involved in generating the data at their clinical research sites. All authors were involved in writing the manuscript and approved it.
-
Funding This study was funded by Roche. F. Hoffmann-La Roche, Ltd (Roche) sponsored the study, participated in the design of the study as well as in the collection, analysis and interpretation of the data. This manuscript was reviewed by Roche, but the decision to submit and publish this manuscript was contingent only on the approval of the lead author and co-authors, including those employed by Roche.
-
Competing interests GRB has received research grants from Roche, Abbott, Pfzier, UCB, Merck Sharp and Dohme and Bristol-Myers Squibb; received consulting fees from Roche, Chugai, Pfizer, UCB and Bristol-Myers Squibb; and was on the speaker bureaux for Roche, Pfizer, Merck Sharp and Dohme, Abbott and Bristol-Myers Squibb. AR-R has received research grants from Roche and Pfizer; received consulting fees from Roche, Chugai, Pfizer, UCB and Merck Sharp and Dohme; and was on the speaker bureaux for Roche and UCB. AC has received research grants from UCB and Pfizer and received consulting fees from Roche, Chugai, Pfizer, UCB, Abbott and Bristol-Myers Squibb. PL has received consulting fees from Roche. MJR has received research grants from Roche. CL has received research grants from Roche, Bristol-Myers Squibb, Pfizer, Human Genome Science, Eli Lilly and Sanofi Aventis and was on the speaker bureau for BMS. EFM has received research grants and consulting fees from Roche and was on the speaker bureau for Roche. PL, LR and MB are employed by Roche. SH, DF and GR have no competing interests to declare.
-
Patient consent Obtained.
-
Ethics approval The study was conducted in accordance with the Declaration of Helsinki and good clinical practice.
-
Provenance and peer review Not commissioned; externally peer reviewed.