Article Text

Abstract
Objectives To investigate the association of lifestyle factors with risk of inflammatory polyarthritis (IP) and rheumatoid arthritis (RA).
Methods The European Prospective Investigation of Cancer, Norfolk, UK (EPIC-Norfolk) gathered lifestyle data from participants aged 40–79 years from 1993 to 1997. Individuals who subsequently developed IP were identified by linkage with the Norfolk Arthritis Register. A Cox proportional hazard model was developed, and a score assigned to each risk factor to calculate the odds of developing IP.
Results 25 455 EPIC participants were followed for a median (IQR) of 14.2 (12.9, 15.3) years; 184 developed incident IP (138 cumulatively fulfilled criteria for RA; 107 were seropositive). Pack-years of smoking were associated with increased risk of IP and RA in men (HR 1.21 (95% CI 1.08 to 1.37) per 10-pack-years) and seropositive IP (HR 1.24 (95% CI 1.10 to 1.41)) for all. Diabetes mellitus was associated with increased risk of IP (HR 2.54 (95% CI 1.26 to 5.09)), while alcohol (HR 0.86 (95% CI 0.74 to 0.99) per unit/day) and higher social class (HR 0.36 (95% CI 0.15 to 0.89) for professionals vs manual workers) were associated with reduced risk. Body mass index was associated with seronegative IP (HR 2.75 (95% CI 1.39 to 5.46) for obese vs normal-weight participants). In women, parity (HR 2.81 (95% CI 1.37 to 5.76) for ≥2 vs no children) was associated with increased risk, and breast feeding (HR 0.66 (95% CI 0.46 to 0.94) for every 52 weeks of breast feeding) was inversely associated with risk. Risk factors from the model were used to generate a ‘risk score’. A total of 1159 (8.4%) women had scores reflecting a >3-fold increased risk of IP over those with a score of 0.
Conclusions Several easily ascertained clinical and lifestyle factors can be used to stratify populations for risk of IP.
- Rheumatoid Arthritis
- Epidemiology
- Smoking
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune disease affecting 0.3–0.8% of the population1–3; however, its aetiology remains an area of intense interest. There have been major advances in our understanding of genetic risk from genome-wide association studies.4 There has also been renewed interest in environmental factors, especially lifestyle factors that are potentially modifiable.5 Smoking is the most consistent association and contributes up to 25% of population attributable risk of RA.6 ,7 The risk appears to be dose-related, stronger in men and in carriers of the shared epitope (SE), and for anti-citrullinated peptide antibody positive (ACPA+) RA.7–11 Some prospective studies also support a protective role of breast feeding in women.12–15 There are data supporting an inverse association with alcohol intake9 ,10 ,16–18 and higher education/social class19–21 and an increased risk with obesity,9 ,22 ,23 but not from prospective studies.24–30 Pregnancy or parity have not been shown to be protective in recent studies,12–15 ,26 ,31–33 although a time-varying decrease in risk from 1 to 5 years postpartum has been postulated.34 We examined these lifestyle factors further in a prospective cohort study, the European Prospective Investigation of Cancer, Norfolk (EPIC-Norfolk), involving adults already being followed for other health outcomes. The EPIC-Norfolk Study has established that several easily ascertained lifestyle factors can predict survival in middle-aged adults.35 We hypothesised that similar factors may also predict the onset of inflammatory polyarthritis (IP). In addition to contributing to a better understanding of the aetiology of IP, such lifestyle factors may also help to stratify subjects in the general population according to their level of risk of IP for further genetic and serological testing, with the aim of preventing the development of IP.
Patients and methods
The study population
The EPIC-Norfolk Study is a population-based, prospective cohort study based in Norfolk, UK. From 1993 to 1997, individuals (99.5% Caucasian) aged 40–79 years from 35 general practice registers were invited to participate. A total of 25 639men and women (33% of those invited) were recruited and provided complete information, details of which are provided elsewhere.36 Briefly, all participants completed a self-administered questionnaire regarding demographic, health and lifestyle factors such as occupation, education, current and lifetime smoking, and physical exercise (see online supplementary material). Hormonal exposures in women, including age of menarche and menopause (if applicable), parity, breast feeding and use of hormone replacement therapy and oral contraceptives, were recorded. Self-reported details of physician-confirmed medical conditions, including depression, cardiovascular disease (CVD), diabetes mellitus (DM) (type 1/2 not specified) and hypertension, were also ascertained. Alcohol consumption (units/week, 1 unit=8 g of alcohol) was derived from a semiquantitative food frequency questionnaire.37 Social class was defined according to the Registrar General's occupation-based classification scheme.38 A validated physical activity index was derived from two questions on past-year work and recreational activities.39 All participants underwent a clinical examination, including measurement of weight, height, blood pressure, and waist and hip circumference, and provided a blood sample. The vital status of all EPIC-Norfolk participants for this study was ascertained up to 31 March 2010 through linkage with the UK Office for National Statistics. No follow-up questionnaires were used in this analysis.
Ascertainment of cases of IP
New cases of IP in the EPIC population were ascertained by linkage with the Norfolk Arthritis Register (NOAR), which covers all the EPIC-Norfolk general practices. Linkage was undertaken in August 2010, and IP cases with symptom onset on or before 31 March 2010 were included. Details of NOAR have also been published elsewhere.40 Briefly, all patients presenting to a general practitioner with IP, defined as inflammation of two or more peripheral joints persisting for at least 4 weeks and onset after 1989, were notified to NOAR. All patients were interviewed and examined by a research nurse to confirm the diagnosis of IP and ascertain fulfilment of the American College of Rheumatology (ACR) 1987 criteria for RA.41 Subjects who fulfilled the above criteria and who were not subsequently given an alternative diagnosis (other than RA, psoriatic or post-viral arthritis) by a rheumatologist were followed. All patients had a baseline blood sample drawn for rheumatoid factor (RF) and ACPA analysis. RF was measured using a particle-enhanced immunoturbidimetric assay where ≥40 IU/ml was considered positive (Orion-Diagnostica). ACPAs were measured using the Axis-Shield CCP2 antigen-plate DIASTAT kit (Axis-Shield, Dundee, UK) where >5 U/ml was considered positive. Ethics approval for both studies was obtained from the Norwich Research Ethics Committee, and all participants gave informed consent.
Outcome measures
The primary outcome measure was development of incident IP. Secondary outcome measures were development of RA and seropositive (RF+ and/or ACPA+) IP. Cases were followed annually in the NOAR study, and RA was ascertained cumulatively by applying 1987 ACR criteria at every visit for up to 5 years.42 It has recently been shown in NOAR that case ascertainment from applying 2010 criteria at baseline is the same as from cumulatively applied 1987 criteria.43
Statistical analysis
Analyses were conducted in Stata V.10. Risk was estimated using a Cox proportional hazards model with the robust option to address heterogeneity of variance. Prevalent cases of IP were excluded. Cases were followed until IP symptom onset; all other participants were censored at time of death, loss to follow-up or 31 March 2010, whichever came first. Time-varying differences were assessed through the proportional hazards test and applied to gender. First, possible risk factors were tested univariately, with adjustment for age and gender. Gender differences were tested through inclusion of interaction terms, and retained only for pack-years of smoking. Pack-years of smoking (every 10 pack-years, adjusted for never being a smoker), units of alcohol (units consumed/day, adjusted for being a teetotaller) and number of years of breast feeding (for women) were modelled as continuous variables. DM, body mass index (BMI) (kg/m2) (as per the WHO definition,44 regrouped as BMI <25 (normal or underweight, referent), BMI 25 to <30 (overweight) and BMI ≥30 (obese or severely obese)), occupational class (regrouped as professionals, non-manual managerial/technical/skilled workers and manual workers), education (regrouped as ‘degree or equivalent’ and ‘no degree’) and parity (for women) were modelled as categorical variables. Variables found to have a trend to significance (p<0.25) on univariate analysis were included in the initial multivariate model. Occupational class rather than education was retained as a measure of socioeconomic status, as this has been shown to be a better discriminator of differentials in mortality in the UK population.45
Results from original data are shown. Results from a sensitivity analysis using multiple imputation for missing values (<5%) were similar (not shown). All results are expressed as HRs with 95% CIs.
Calculation of incidence rates and development of a ‘risk score’
We calculated the cumulative 10-year incidence of IP from the number of incident cases per person-year of follow-up. We then calculated a risk score based on the β coefficients from our IP models (with negative scores for protective factors), and the odds of developing IP based on the score by logistic regression. We performed internal validation via bootstrapping (not shown), which did not materially affect the CIs of the estimates we have reported.
Results
After exclusion of 180 prevalent cases, 25 455EPIC participants remained for analysis. The median (IQR) age was 58.9 (50.9, 66.9) years, 45.4% were men, and the median (IQR) duration of follow-up was 14.2 (12.9, 15.3) years. During 342 916 person-years of follow-up, 184 participants (128 (69.6%) women) developed incident IP, of which 138 cumulatively fulfilled criteria for RA; 57.6% and 35.9% were, respectively, RF and ACPA positive (60.4% seropositive). The median (IQR) time to onset of IP was 62.7 (27.8, 104.0) months, and the median (IQR) age at IP onset was 65.2 (57.6, 73.2) years. Table 1 shows the baseline characteristics of cases and the unaffected cohort. Patients with IP were more likely to be current smokers (19.6% vs 12.1% for men, 15.9% vs 11.3% for women). Among male ever smokers, patients were more likely to be heavy smokers (median (IQR) 26.6 (10.1, 50.0) vs 19.0 (9.5, 32) pack-years, p=0.03). Female smokers, on average, smoked less than male smokers (median (IQR) 9.3 (5.0, 23.1) pack-years for women with IP vs 26.6 (10.1, 50.0) for men with IP). Women who developed IP were more likely than those without IP to be obese (25.8% vs 16.8%, p=0.01), have DM (6.3% vs 1.5%, p<0.001), be of a non-manual occupational class (48.8% vs 61.5%, p=0.003) and have at least two children (83.6% vs 71.5%, p=0.01), but breast fed for a shorter time (median (IQR) 5 (0, 28) vs 10 (1, 36) weeks, p=0.02).
Baseline characteristics of inflammatory polyarthritis (IP) cases versus non-cases by gender
The risk of developing IP, RA and seropositive IP associated with each risk factor are presented in tables 2⇓–4, respectively. Two models were developed: one for the whole cohort, and one for women only. Smoking was associated with a dose-dependent linear 20% increase in IP risk for every 10 pack-years smoked in men (adjusted HR 1.21 (1.08 to 1.37)), but not in women (adjusted HR 1.00 (0.82 to 1.21)). A similar trend was seen for RA; however, risk of seropositive IP was increased in both genders (adjusted HR 1.24 (1.10 to 1.41)). When analysed by smoking status, female current versus non-smokers were at about a 50% increased risk of IP, RA and seropositive IP. Alcohol appeared protective for the development of IP (adjusted HR 0.86 (0.74 to 0.99)), RA and seropositive IP, with a 14% risk reduction per unit consumed per day. Higher BMI showed a trend towards association with risk of IP (adjusted HR 1.45 (0.95 to 2.21) for BMI ≥ 30 vs normal weight) and RA, but not seropositive IP (HR 1.05 (0.61 to 1.79), age- and gender-adjusted). A post hoc analysis looking at seronegative IP revealed a nearly threefold increase in risk (HR 2.75 (1.39 to 5.46) for BMI ≥ 30 vs normal weight, age- and gender-adjusted). Self-reported DM was associated with an increased risk of IP, especially in women (adjusted HR 4.28 (2.04 to 9.01)), which was independent of BMI. Higher occupational class or degree education was associated with reduced risk of IP. In women, having two or more children was associated with a doubling of risk (adjusted HR 2.81 (1.37 to 5.76) vs nulliparous), while breast feeding (adjusted HR 0.66 (0.46 to 0.94) per year of breast feeding) showed a dose-dependent inverse association with IP, RA and seropositive IP.
Predictors of inflammatory polyarthritis
Predictors of rheumatoid arthritis (ACR 1987 criteria)
Predictors of seropositive (RF+ or ACPA+) inflammatory polyarthritis
Calculation of incidence and development of risk score
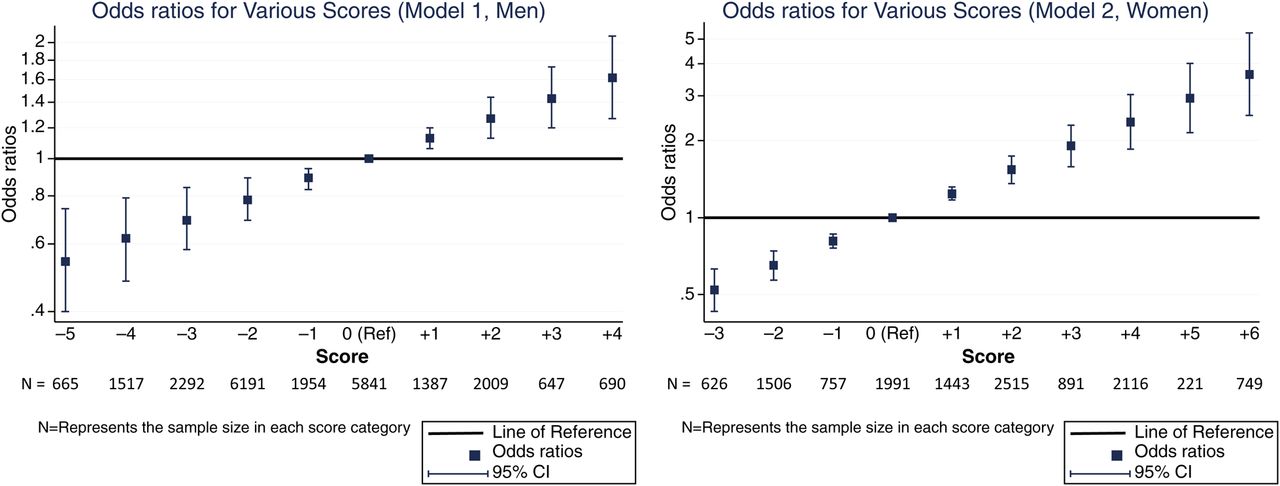
The 10-year cumulative incidence of IP was 0.37% in men and 0.67% in women. We devised a risk score based on the β coefficients from our IP models (table 5). Continuous variables were assigned points for each unit of measurement up to a maximum which was within the range for our cohort. For men, every additional 10 pack-years of smoking (up to a maximum of 4 points for >30 pack-years), being obese and having DM were scored positively, whereas drinking up to 3 units of alcohol per day (1 point for each whole unit, up to a maximum of 3 points) and being of a higher occupational class were scored negatively. For women, ‘current’ smoking replaced pack-years smoked, and additional scores were assigned for having ≥2 children (positive) and every additional 6 months of breast feeding (negative, up to a maximum of 4 points for ≥2 years). The total score is the sum of all individual scores, and a higher total score implies a higher risk of IP. We derived relative estimates of risk at each score by logistic regression (figure 1, table S1). For example, the risk for men with a score of 5 was approximately double the risk of men who scored 0 (area under receiver operating characteristics curve (AUC)=0.59). The model performed better in women (AUC=0.66). For example, a score of 5 points implied an approximately threefold increased risk of IP, which translates to a 10-year cumulative incidence of >2%. Scores ≥5 were seen in 1159 (8.4%) women in our cohort.
Assignment of risk score
{kind=link}
Odds ratios for various scores.
Discussion
In this large population-based prospective cohort, several lifestyle factors were associated with risk of IP. These factors, which are easily ascertained in primary care, can be combined to develop a simple screening tool to identify individuals with an up to sixfold increased risk of IP compared with the population, who could then be targeted for more focused risk assessments. Some of the risk factors we found to be associated with IP are consistent with previous literature and may suggest potential pathogenic pathways. Smoking is thought to interact with the SE to increase the risk of ACPA+ RA.8 ,10 Previous literature has noted a weaker association in women11 and a threshold dose of 10–20 pack-years before the increased risk is apparent.6 Most women in our cohort had smoked less than this, which explains the lack of association between smoking dose and IP risk in women in our cohort. Alcohol has previously been shown to be protective in case–control studies9 ,10 ,16 ,17 and one prospective study of women.18 We have shown that the negative association also holds true for men. Biologically, this may be the result of antioxidant/anti-inflammatory mechanisms including reduction of postprandial oxidative stress,46 increased urate production, antioxidant properties from polyphenolic flavonoids,47 or downregulation of the immune response.48 Others have reported that alcohol is particularly protective in smokers,17 but we found no significant interaction with smoking in our study (data not shown). The association with BMI is also of interest. Obesity has been found to be a risk factor for IP in case–control studies,9 ,22 ,23 but not in cohort studies.25 ,26 ,28 ,29 We have previously noted a time-varying risk from obesity in this cohort.30 In this larger study, we also noted a differential risk by serotype. Obesity markedly increased the risk of seronegative but not seropositive IP, which supports previous literature.9 The effect of obesity may be mediated through increased availability of oestrogen, or through an altered relationship between leptin and adiponectin contributing to a pro-inflammatory state akin to that noted in CVD.49–51 Adiponectin levels are also decreased in type 2 DM, and are inversely related to levels of circulating tumour necrosis factor α. The resulting proinflammatory milieu may also explain the strong association observed between IP and DM in our study.52
In women, parity of ≥2 was associated with a doubling of risk for IP in our study. Most previous studies have shown either a decreased risk34 ,53 ,54 or no association with parity.12–15 ,26 ,31–33 The apparent contradiction may be due to our relatively older cohort. Episodes of pregnancy may initially have ‘protected’ these women against RA and this ‘protection’ may have waned over time and simply postponed the onset of IP rather than truly reducing risk. This time-varying protective effect has been previously suggested.34 Conversely, breast feeding was associated with a dose-dependent reduction in risk when adjusted for parity. This is consistent with previously published literature.12–15 However, the negative association after what would be many years since cessation of breast feeding is difficult to explain. It may again be an example of ‘depletion of susceptibles’ in that a surge of (proinflammatory) prolactin during the first breastfeeding episode may have unmasked latent RA in susceptible individuals at an early age before their entry into our cohort.55
We have therefore confirmed that there are a number of lifestyle factors that can influence the risk of IP. We then attempted to develop a risk score for IP. In developing this, we specifically limited our model to variables easily ascertained at a routine primary care consultation using only pack-years of smoking, alcohol consumption, occupational class, BMI and presence of DM in men, and smoking status, alcohol consumption, occupational class, BMI, presence of DM, parity and duration of breast feeding in women. Some of these factors already form part of the lifestyle advice given for CVD and cancer prevention. With this model, we could identify a number of individuals who were up to six times more likely than the background population to develop IP. Although the absolute risk is small, this model, if validated, would provide a simple population/primary care screen for stratifying populations for more detailed risk assessment approaches such as serological testing or genetic screening. For example, others have noted that persons with two or more first-degree relatives with RA and positive ACPA have a high risk of developing RA over 5 years.56 Our simple screen would complement such an approach at the population level, first by helping to target modifiable lifestyle factors such as BMI and smoking, but also allowing enhanced screening for ACPA and at-risk genotypes in a higher risk population for potential pharmacological interventions.
Our study has several strengths. Most importantly, these data were derived from a large prospective population-based cohort, and only incident cases were included, so our results are not subject to selection or recall bias. Cases of IP/RA were ascertained by examination by study personnel rather than through self-reported questionnaires or medical record review, and all cases were followed-up and ACR 1987 criteria for RA were applied cumulatively. There are, however, several limitations. Lifestyle data were self-reported and collected cross-sectionally at a single time point, and there may have been increasing misclassification later into the follow-up period. Although every attempt was made to include all incident cases of IP through general practices and speciality rheumatology clinics, there may be incomplete case ascertainment, leading to an underestimation of IP incidence. Also, we were limited to studying risk factors included in the EPIC-Norfolk questionnaire, and hence important predictors such as family history (a clinical surrogate for genetic risk) and occupational exposure (eg, silica) could not be included in our model.57–59 Our cohort comprised late-middle-aged individuals, and IP cases of earlier onset would have been excluded. Nevertheless, the peak age of onset of IP is in the 6th and later decades,42 so these data are generalisable to most patients with IP and are also of relevance, as they include the population in which active screening programmes (eg, for CVD) already exist. Lastly, we have based this risk score on just 184 cases, and hence the model will need to be validated in other large cohorts. In spite of including several lifestyle factors in our model, the overall attributable risk from these is likely to be small, as RA has a strong genetic contribution to its development. As expected, the AUCs for our models were marginal.
Future work should involve combining genetic and environmental data in the same cohorts to characterise gene–environment interactions in the development of IP/RA. Studying these within the paradigm of seropositive and seronegative disease, as has been previously suggested, will be crucial to advancing our understanding of the disease.
Acknowledgments
NOAR is funded by Arthritis Research UK, Chesterfield, UK (Grant reference 17552); EPIC-Norfolk is funded by The European Commission ‘Europe against Cancer’ Programme, Cancer Research UK, Medical Research Council with additional support from the Stroke Association, British Heart Foundation, Department of Health, Food Standards Agency and the Wellcome Trust. This report includes independent research supported by the National Institute for Health Research Biomedical Research Unit Funding Scheme. The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
-
Handling editor Tore K Kvien
-
Contributors All authors contributed to this paper and fulfil the criteria for authorship.
-
Funding NOAR is funded by Arthritis Research UK, Chesterfield, UK (Grant reference 17552); EPIC-Norfolk is funded by The European Commission ‘Europe against Cancer’ Programme, Cancer Research UK, Medical Research Council with additional support from the Stroke Association, British Heart Foundation, Department of Health, Food Standards Agency and the Wellcome Trust.
-
Competing interests None.
-
Ethics approval Norwich Research Ethics Committee.
-
Patient consent Obtained.
-
Provenance and peer review Not commissioned; externally peer reviewed.