Article Text

Abstract
Objective To determine the efficacy and safety of ocrelizumab (OCR) with methotrexate (MTX) in MTX-naive rheumatoid arthritis (RA) patients.
Methods In a randomised, double-blind, controlled trial, patients received placebo+MTX (MTX; n=210), OCR 200 mg×2+MTX (OCR 200; n=200) or OCR 500 mg×2+MTX (OCR 500; n=203). OCR/placebo (two intravenous infusions) was given on days 1 and 15, with fixed re-treatment scheduled at weeks 24/26, 52/54 and 76/78. Due to early termination of OCR dosing, there was no formal primary end point analysis (change from baseline in modified total Sharp score (ΔmTSS) at week 104). Analyses are reported for week 52 outcomes.
Results At week 52, treatment with OCR+MTX compared with MTX alone reduced progression of joint damage (mean (SD) change in ΔmTSS: OCR 200, 0.66 (4.51); OCR 500, 0.27 (2.91); MTX alone, 1.59 (4.82); p=0.001 and p=0.003, respectively vs MTX alone) and improved clinical signs and symptoms (American College of Rheumatology 20 response: OCR 200, 73.0%; OCR 500, 71.0%; MTX alone, 57.5%; p<0.005 for each OCR vs MTX alone). Serious infection rates per 100 patient-years were similar with OCR 200 and MTX alone (2.6 (95% CI 0.9 to 6.1) and 3.0 (1.1 to 6.5), respectively), but higher with OCR 500 (7.1 (3.9 to 11.9)).
Conclusions OCR 200 mg and 500 mg with MTX in MTX-naive patients with RA were effective in inhibiting joint damage progression and improving RA signs and symptoms. OCR 500 mg with MTX was associated with an increased rate of serious infections.
This paper is freely available online under the BMJ Journals unlocked scheme, see http://ard.bmj.com/info/unlocked.dtl
Statistics from Altmetric.com
Introduction
Early treatment for rheumatoid arthritis (RA) with disease-modifying antirheumatic drugs (DMARDs) such as methotrexate (MTX), either alone or in combination with biological agents, effectively controls disease activity and prevents joint damage in patients with RA.1,–,3 Rituximab, a murine–human chimeric monoclonal antibody that selectively targets B cell-surface CD20, in combination with MTX, is efficacious in patients with active RA who have an inadequate response to DMARDs and tumour necrosis factor (TNF) inhibitor therapies4,–,6 and in patients with early disease naive to MTX.7
Ocrelizumab (OCR) (rhuMAb 2H7) is a humanised monoclonal antibody that targets CD20+ B cells. OCR is characterised by enhanced antibody-dependent cell-mediated cytotoxicity and reduced complement-dependent cytotoxicity compared with rituximab in vitro (Roche, data on file). The clinical significance of these differences is currently unclear.
A phase I/II study, ACTION (A randomized, placebo-ConTrolled, blinded, phase I/II study of escalatIng doses of Ocrelizumab in patients with moderate to severe RA on stable doses of coNcomitant MTX), demonstrated that OCR plus MTX was well tolerated in patients with RA. Best clinical responses and low immunogenicity were observed at doses of ≥200 mg when administered as two infusions, 2 weeks apart.8 Accordingly, two doses of OCR, 200 mg and 500 mg, were selected for further phase III investigation.
The phase III FILM (SaFety and effIcacy of ocreLizumab in combination with Methotrexate (MTX) in MTX-naive subjects with rheumatoid arthritis) study was originally designed with a 104-week, double-blind treatment period in MTX-naive patients with RA. OCR development in RA was terminated by the sponsors before all patients reached the primary end point of the study at 104 weeks as a result of an overall risk/benefit assessment based on the two pivotal phase III RA trials, STAGE (STudy to evaluate the safety and efficacy of ocrelizumab compared to placebo in patients with Active rheumatoid arthritis continuinG mEthotrexate treatment) (in MTX inadequate responders)9 and SCRIPT (Study to evaluate the safety and efficacy of oCRelIzumab compared to Placebo in patients with active rheumatoid arthritis who have had an inadequate response to at least one anti-TNF therapy) (in TNF inadequate responders).10 However, all patients in the current FILM trial had received ≥2 courses of treatment and completed 52 weeks of double-blind, placebo-controlled treatment. Clinically relevant safety and efficacy results up to 52 weeks are presented here.
Methods
Patients
Patients (≥18 years old) had active, moderate-to-severe RA (according to the revised 1987 American College of Rheumatology (ACR) criteria) for ≥3 months but <5 years; swollen joint count ≥8 (66 joint count) and tender joint count ≥8 (68 joint count) at screening and baseline; C reactive protein (CRP) levels ≥1.0 mg/dl at screening; and were seropositive for rheumatoid factor (RF) and/or anticitrullinated peptide antibody (ACPA). Patients had not received MTX or any biologic for RA previously and were candidates for MTX therapy. All prior DMARD therapy was discontinued ≥4 weeks before baseline (12 weeks for leflunomide).
The main exclusion criteria were rheumatic autoimmune diseases or inflammatory joint disease other than RA, including significant systemic involvement secondary to RA; functional Class IV RA (ACR criteria); congestive heart failure (New York Heart AssociationClass III and IV); or severe chronic obstructive pulmonary disease (forced expiratory volume in one second <50% predicted). All patients were screened for tuberculosis (TB) according to local/national guidelines. Patients with active TB or chronic active hepatitis (Hep) B or C were excluded. Patients receiving treatment for latent TB infection were eligible. Those who were HepB core antibody positive but HepB surface antigen negative and HepB viral DNA negative were eligible, but monitored for HepB viral DNA.
Study design
FILM was a randomised, double-blind, parallel-group, placebo-controlled phase III study (figure 1A) conducted at 147 centres in 21 countries, with approximately half of patients recruited in the USA. Recruitment occurred between 27 June 2007 and 19 December 2008.
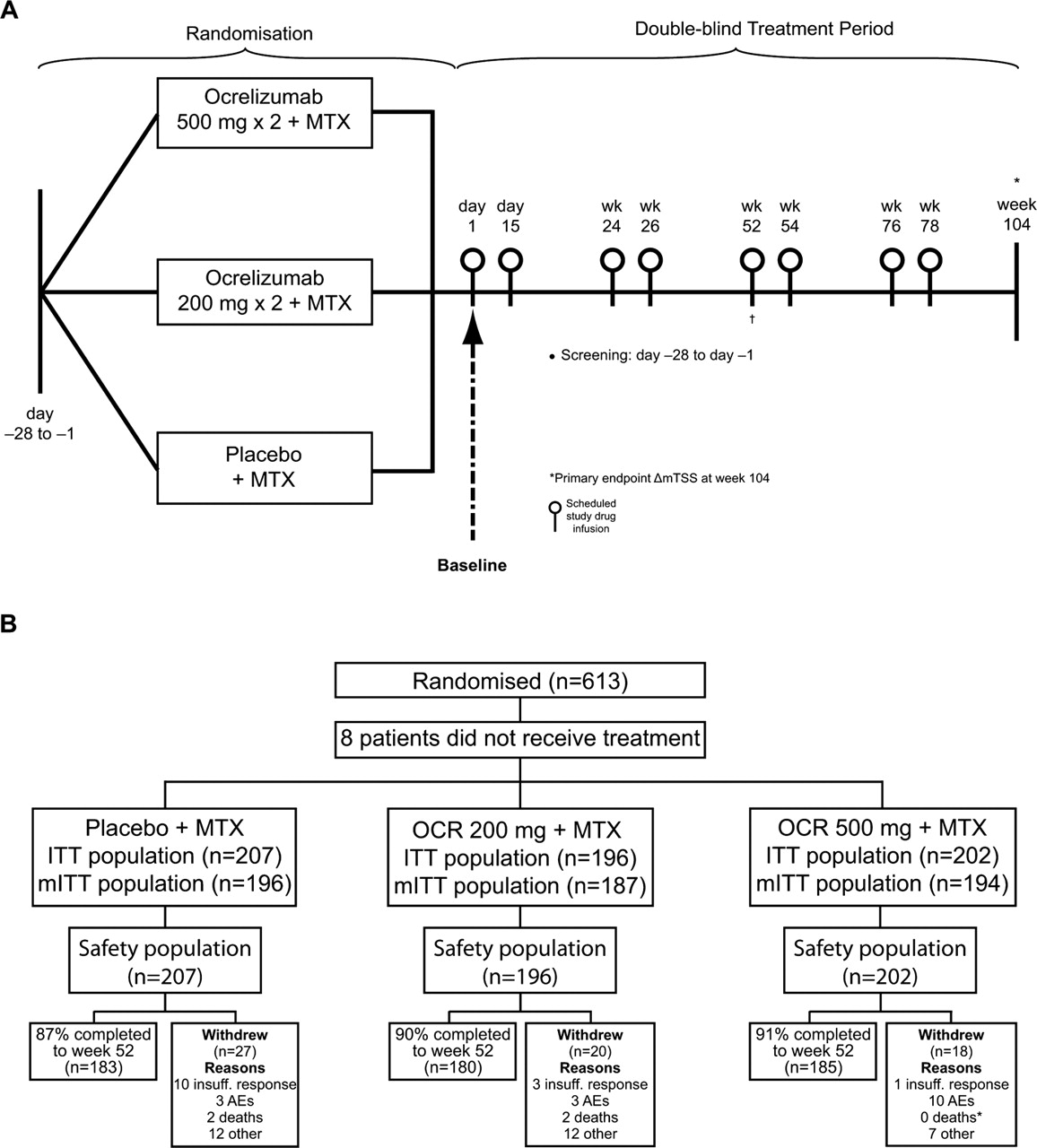
(A) Study design. (B) Disposition of patients. *One patient withdrew from the study due to an AE. The patient died; the cause of death was ischaemic cerebral infarction. †Outcomes reported at week 52. AE, adverse event; ITT, intent-to-treat; mITT, modified ITT; MTX, methotrexate; OCR, ocrelizumab.
Patients were randomised (1:1:1) to placebo×2+MTX (PLB/MTX), OCR 200 mg×2+MTX (OCR200/MTX) or OCR 500 mg×2+MTX (OCR500/MTX) using a randomised interactive voice recognition system. Randomisation was stratified by region (USA or rest of world) and CRP status (≤3.0 or >3.0 mg/dl). Patients received two intravenous infusions separated by 14 days of OCR or placebo on days 1 and 15 (Course 1) and on weeks 24 and 26 (Course 2). Patients were scheduled to receive further infusions at weeks 52 and 54 and at weeks 76 and 78 but most did not due to termination of the study. All patients were started on MTX on day 1 at an initial dose of 7.5 mg/week, increasing to 20 mg/week by week 8, as tolerated. Intravenous methylprednisolone 100 mg was administered 30 min before each infusion. Paracetamol (1 g) and antihistamine (diphenhydramine HCl 50 mg or equivalent) were recommended as additional premedications. Any rescue medication for RA could be administered at any time at the discretion of the investigator. Such patients could remain in the study but were considered non-responders for statistical efficacy analyses after initiation of rescue therapy.
Scheduled radiographs of the hands and feet were obtained at baseline (within±7 days of day 1) and at weeks 24 and 52 (and before rescue and at time of early withdrawal). Radiograph joint scoring (blinded) was done centrally by two independent joint assessors at a reading centre according to a detailed x-ray reading charter.
The study was conducted in accordance with the ethical principles of the Declaration of Helsinki. Ethical approval from the local institutional review board was obtained before the start of the study and all patients provided written informed consent. This study is registered at ClinicalTrials.gov, number NCT00485589.
After patients completed the 52-week treatment period, they transitioned into a safety follow-up period that included B cell monitoring.
Assessments
Efficacy
The prespecified primary end point was change from baseline in the van der Heijde-modified total Sharp score (ΔmTSS) at week 104. Due to early termination of dosing, formal analysis of the primary end point was not feasible. However, given the robust nature and the clinical significance of these data, ΔmTSS at week 52 was identified as the key efficacy end point. Additional week 52 efficacy endpoint analyses included the proportions of patients without radiographic progression and the proportion of patients achieving ACR20/50/70 responses.11 Other clinical end points included Health Assessment Questionnaire Disability Index improvement, Disease Activity Score in 28 joints erythrocyte sedimentation rate (DAS28-ESR), and proportion of patients achieving DAS28 remission (DAS28<2.6) and low disease activity (LDA; DAS28≤3.2).
Pharmacodynamics
B cell (CD19+) counts in blood samples were determined using fluorescent activated cell sorter analysis at screening, baseline and weeks 2, 4, 12, 24, 26 and 52.
Safety
Adverse events (AEs) were recorded throughout the study with intensities graded according to the National Cancer Institute Common Terminology Criteria for AEs, Version 3. Safety data through week 52 are reported.
Statistical analysis
All statistical analyses were performed on data collected through each patient's week 52 visit. This study was considered to be supportive and not confirmatory following the early termination of treatment, and therefore no correction for type I error was deemed necessary. The sample size of 200 patients per treatment group was designed to provide at least 80% power to detect a difference in the distributions of ΔmTSS at week 104 between the OCR and PLB/MTX groups using the Wilcoxon test, with a 0.025 two-sided significance level. The calculation was also based on a 0.60 probability that ΔmTSS would be less in patients receiving OCR than in patients receiving PLB/MTX.
Clinical efficacy analysis was performed using the intent-to-treat (ITT) population, which included all patients who received any study treatment. Two patients with missing baseline CRP data were excluded from all stratified efficacy analysis. The safety population comprised all patients who received any study treatment and at least one safety assessment. Radiographic end points were assessed using a modified ITT (mITT) population, which included patients in the ITT population who had both a baseline radiograph and at least one postbaseline radiograph. All efficacy analyses were stratified by region and by CRP status.
Group comparisons for ΔmTSS at week 52 were made using a non-parametric test statistic (Van Elteren) stratified for region and CRP status. For any patient receiving rescue therapy before week 52, the mTSS score was set to ‘missing’ 8 weeks after rescue therapy was received. Missing mTSS data imputations were applied afterwards, primarily using linear extrapolation. The proportion of patients without radiographic progression (ΔmTSS≤0) at week 52 was analysed using the Cochran–Mantel–Haenszel (CMH) test, and the primary analysis method used observed data for mTSS. Any patient with missing data at the analysis time point was classified as having radiographic progression.
The proportions of patients achieving ACR20, ACR50 or ACR70 responses at week 52 were analysed using a CMH test statistic for comparing group differences. Results were expressed as proportions with an adjusted 95% CI for the treatment difference and corresponding CMH p value. Early withdrawals or those who received rescue medication were considered non-responders.
Results
Patient disposition and baseline characteristics
Patient disposition is shown in figure 1B. A total of 613 patients were randomised (PLB/MTX, n=210; OCR200/MTX, n=200; OCR500/MTX, n=203). Eight patients (n=3, n=4 and n=1, respectively) from the randomised population did not receive any treatment and were excluded from the analysis populations. The safety and ITT populations comprised 605 patients. Twenty-eight patients in the ITT population were excluded from the mITT population due to missing baseline or other postbaseline radiograph data; the mITT population comprised 577 patients.
Most patients (93.2%, 93.4% and 92.1% in the PLB/MTX, OCR200/MTX and OCR500/MTX groups, respectively) received the scheduled two courses of medication prior to week 52. A total of 65 patients (27 (12.9%), 20 (10.0%) and 18 (8.9%), respectively) withdrew before week 52, and 38 patients (22 (10.5%), 8 (4.0%), and 8 (3.9%), respectively) received rescue medication. Approximately 50% of patients who received rescue therapy remained in the study through week 52.
Demographics and baseline characteristics were well balanced across groups (table 1).
Baseline demographics and disease characteristics (ITT population)
Radiographic analysis
Differences in distributions of ΔmTSS for OCR200/MTX and OCR500/MTX compared with PLB/MTX were statistically significant at week 52 (p=0.0010 and p=0.0033, respectively) (figure 2A). Mean changes from baseline were 0.66 (SD, 4.51) (median 0.00, IQR 0.00, 0.50)) for OCR200/MTX, 0.27 (2.91) (median 0.00 (IQR 0.00, 0.50)) for OCR500/MTX and 1.59 (4.82) (median 0.00 (IQR 0.00, 1.50)) for PLB/MTX, corresponding to inhibition of mean radiographic joint damage progression of 58% and 83% for OCR200/MTX and OCR500/MTX, respectively.

Radiographic outcomes at 52 weeks. (A) Changes in mTSS. All comparisons are to placebo using Van Elteren's test stratified by region and screening CRP status. Missing data were imputed using linear extrapolation. *p<0.05; **p≤0.001. (B) Proportion of patients with no radiological progression. Cochran–Mantel–Haenszel analysis was used to calculate p values. Analysis was stratified by region and screening CRP status. *p<0.05; **p≤0.001. (C) Cumulative distribution plot of change from baseline in mean change in mTSS. Missing data were imputed using linear extrapolation. CRP, C reactive protein; mTSS, van der Heijde-modified total Sharp score; MTX, methotrexate; OCR, ocrelizumab.
The proportions of patients without radiographic progression at week 52 were significantly higher for OCR200/MTX (66.3%, p=0.0030) and OCR500/MTX (68.8%, p=0.0006) compared with PLB/MTX (51.0%) (figure 2B). A cumulative distribution plot of change from baseline at week 52 also supports this observation (figure 2C).
Overall and split by treatment group, there did not appear to be a difference in the mean change from baseline in DAS28 at week 52 between patients with and without mTSS progression (data not shown).
Signs and symptoms
At week 52, the proportions of patients achieving ACR20/50/70 responses in the OCR200/MTX and OCR500/MTX groups were statistically significantly different from the PLB/MTX group (figure 3A). The proportions of patients achieving DAS28-ESR remission at week 52 were also significantly higher in both OCR groups relative to PLB/MTX (p<0.0001 for each comparison) (figure 3B). Similarly, significant differences between OCR and PLB/MTX were also observed for DAS28-ESR LDA at week 52 (p<0.0001 for each comparison) (figure 3C).

{kind=link}
{kind=link}
{kind=link}
Clinical efficacy at 52 weeks. 95% CIs indicated. Cochran–Mantel–Haenszel analysis was used to calculate p values. Analysis was stratified by region and screening CRP status. Last observation carried forward to withdrawal or rescue for joint counts. *p<0.05; **p≤0.001; ***p≤0.0001. (A) ACR20/50/70 response rates. Missing change from baseline in CRP substituted with change from baseline in ESR. Observed data for other ACR components until withdrawal/rescue. (B) DAS28-ESR remission. (C) Proportion of patients with low Disease Activity Score (DAS28-ESR). Observed data for visual analogue scale and ESR until withdrawal/rescue. ACR, American College of Rheumatology; CRP, C reactive protein; DAS28-ESR, Disease Activity Score in 28 joints erythrocyte sedimentation rate; MTX, methotrexate; OCR, ocrelizumab.
The clinical responses were accompanied by decreases in autoantibody levels; the mean decreases in RF and anticitrullinated peptide antibody levels from baseline to week 52 were 73% and 60%, respectively, for OCR200/MTX and 67% and 60%, respectively, for OCR500/MTX compared with 36% and 10%, respectively, for PLB/MTX.
The proportion of patients achieving a clinically meaningful reduction (defined as ≥0.25 units) in the Health Assessment Questionnaire Disability Index score at week 52 was 73.0% (95% CI 67% to 79%) for OCR200/MTX and 72.5% (95% CI 66% to 79%) for OCR500/MTX versus 64.3% (95% CI 58% to 71%) for PLB/MTX (p=0.0393 and p=0.0763, respectively).
Pharmacodynamics
A rapid reduction of peripheral CD19+ B cells to low levels was observed consistently in all patients in both OCR groups by week 2. At weeks 24 and 52, mean (median) CD19+ B cell counts (cells/µl) for OCR500/MTX were 13.7 (6) and 17.6 (5), respectively. For OCR200/MTX, the CD19+ B cell counts were 21.8 (10) and 27.7 (10) at weeks 24 and 52, respectively, suggesting a somewhat faster repletion of peripheral B cells in the low dose group. At week 52, B cell levels had not returned to predepletion levels in 89.3% and 94.8% of the OCR200/MTX and OCR500/MTX groups, respectively.
Safety
The overall incidence of AEs was similar across the treatment groups over 52 weeks (table 2). The most common AEs (all treatment groups combined) were infusion-related reactions (IRRs), upper respiratory tract infections, hepatotoxicity and nausea. The incidence of serious AEs (SAEs) was higher for OCR500/MTX (13.9%) compared with OCR200/MTX (9.2%) and PLB/MTX (10.1%), with infections and infestations being the most frequently reported SAEs overall. IRRs occurred more frequently for OCR200/MTX (26.5%) and OCR500/MTX (26.7%) than for PLB/MTX (8.7%). The incidence of IRRs was markedly reduced with the second infusion and the second course. Only one case of a serious IRR (Grade 3) was reported (OCR500/MTX).
Summary of AEs* reported over 52 weeks (safety population)
Overall, infections were reported with similar frequency across the treatment groups (table 2). However, the incidence of serious infections was numerically higher for OCR500/MTX (5.0%) versus OCR200/MTX (2.6%) and PLB/MTX (2.9%). The serious infection rate per 100 patient-years was also higher for OCR500/MTX (7.1 (95% CI 3.9 to 11.9)) versus OCR200/MTX (2.6 (0.9 to 6.1)) and PLB/MTX (3.0 (1.1 to 6.5)) (table 3). Respiratory infections (pneumonia and bronchitis) were the most common type of serious infection. A post hoc analysis by region showed that serious infection rates were substantially higher with OCR500/MTX among patients from the Asia-Pacific region (n=182; Korea, Philippines, Taiwan and Thailand) than among patients from the rest of the world (30.1 vs 4.5 per 100 patient-years). This disparity was not observed in the OCR200/MTX or PLB/MTX treatment groups.
Serious infection rates (safety population)
Three opportunistic infections were reported during the 52-week treatment period; all occurred in patients receiving concomitant corticosteroids. A histoplasmosis infection occurred in one patient (OCR200/MTX) in an endemic area in the midwestern USA. A systemic candida infection was seen in a patient from South Korea (OCR500/MTX) who was treated with high-dose corticosteroids for pulmonary alveolar haemorrhage. A second patient from South Korea (OCR500/MTX) presented with varicella pneumonia while hospitalised for interstitial lung disease. All three events resolved without sequelae. No cases of progressive multifocal leucoencephalopathy were reported.
Malignancies were reported in three patients: prostate cancer (PLB/MTX) and squamous cell carcinoma of the skin (OCR500/MTX) resulting in discontinuation of study drug and thyroid cancer (PLB/MTX).
A total of 67 patients across the three treatment groups were HepBcAb positive at screening; no clinical evidence of hepatitis reactivation was observed during the study. Ninety-four patients had a positive purified protein derivative or Quantiferon test at baseline; none of these patients developed active TB. Twenty-two of these patients were receiving prophylactic treatment for latent TB that had started prior to baseline and continued into the double-blind treatment period.
Five patients died during the 52-week study period: two receiving PLB/MTX (one patient (female, 62) with concomitant hypertension and a history of type B aortic dissection died from acute myocardial infarction and one patient (male, 53) with concomitant peripheral oedema died from congestive heart failure), two receiving OCR200/MTX (one patient (female, 74) from haemorrhagic stroke, one patient (female, 60) with concomitant hypertension and chronic venous insufficiency died from acute respiratory failure) and one receiving OCR500/MTX (female, 63, with concomitant paroxysmal atrial fibrillation, hypertension, diabetes, atherosclerosis and chronic bronchitis died from ischaemic cerebral infarction).
Reductions were seen in mean immunoglobulin (Ig) A, IgG and IgM values following the first infusion of study drug in all three treatment groups. Following these initial reductions, mean Ig levels stabilised from approximately week 12 to week 52 in the PLB/MTX group, while continued reductions were seen after the second infusion of study treatment in the OCR200/MTX and OCR500/MTX groups. Mean IgA, IgG and IgM values were lower at week 52 in the two OCR/MTX treatment groups compared with the PLB/MTX group. Mean values, however, remained within the normal ranges for all three classes of immunoglobulins. IgM levels were below the lower limit of normal in two patients with serious infections at the time of event, both in the OCR500/MTX group and both from South Korea. One patient experienced varicella pneumonia, the other patient a systemic candida infection; both events resolved without sequelae. All other serious infections were not associated with IgG, IgM or IgA levels below the lower limit of normal around the time of event.
Discussion
OCR dosing in the FILM study was terminated prematurely following the sponsor's reassessment of the benefit/risk of B cell depleting agents, including rituximab, for treatment of MTX-naive patients with early RA who have a wide range of therapeutic options available to them. This prevented assessment of the protocol-specified primary end point of radiographic progression at 104 weeks. Nevertheless, analyses of data from the 52-week double-blind, placebo-controlled treatment period provide useful insights of general interest on the efficacy and safety of OCR+MTX versus MTX alone in MTX-naive patients with RA.
OCR with MTX substantially inhibited joint damage progression in early RA. At week 52, each dose of OCR significantly reduced mean ΔmTSS and prevented radiographic progression in a greater proportion of patients versus MTX alone. These data are similar to those observed for rituximab in MTX-naive patients with RA7 and in two related OCR phase III trials, STAGE and SCRIPT. In STAGE, OCR 200 mg and 500 mg each significantly inhibited radiographic progression over 48 weeks versus placebo in patients with an inadequate response to MTX.9 In SCRIPT, which studied a more refractory TNF-IR population, only OCR 500 mg significantly inhibited structural damage progression.10 With regard to the 500 mg dose, the FILM data are consistent with other reports of biologics in MTX-naive populations.1,–,3
OCR with MTX also improved the signs and symptoms of RA, as assessed by ACR and DAS28 response rates. A higher proportion of patients in the OCR groups than in the placebo group achieved high-hurdle end points such as DAS28 remission and LDA. This improvement was maintained over 52 weeks. The ACR response rates were comparable with those observed in a similar population treated with rituximab.7
The overall rate of AEs was similar among the treatment groups. However, patients in the OCR500/MTX group experienced a higher rate of SAEs. The most common AEs were IRRs and infections. Mild-to-moderate IRRs (Grade 1 or 2) were reported in approximately a quarter of OCR patients. As observed with rituximab12 and in other OCR RA studies8,–,10 the incidence of IRRs was the highest during the first infusion of the first treatment course, before declining with subsequent infusions and courses.
The serious infection rate with OCR200/MTX was similar to that observed with PLB/MTX, whereas a threefold higher rate was observed with OCR500/MTX, consistent with results from other phase III trials of OCR in RA. A subgroup analysis suggested that patients in the 500 mg group recruited from the Asia-Pacific region had a higher rate of serious infections. This imbalance largely, but not entirely, drove differences in serious infection rates compared with the other two study groups. Similar trends were observed in the SCRIPT trial, where a higher rate of serious infections appeared to be driven largely by patients from Japan.10
Both doses of OCR rapidly depleted B cells shortly after infusion. However, there were differences with regard to B cell repletion at week 24 and again at week 52 such that there appeared to be faster repletion in the peripheral blood in the lower dose group. While those differences between the two OCR groups were small, they were remarkably consistent throughout all phase III trials conducted with OCR. Because initial peripheral B cell depletion is profound and consistent in all patients, no correlation between peripheral B cell counts and response rates can be established. However, it could be hypothesised that subtle differences in peripheral B cell counts translate into much greater differences in secondary lymphoid tissues (eg, spleen, lymph nodes), as well as the synovium. This could help explain differences between the two doses with regard to efficacy, particularly the radiographic results, and potentially differences in safety, especially infections. No reliable biomarker of tissue B cell depletion in humans is available to date.
In conclusion, 52-week results from FILM indicate that OCR200/MTX in combination with MTX was effective and well tolerated in MTX-naive seropositive patients with RA. OCR500/MTX was associated with an increased risk of serious infections, with patients recruited in the Asia-Pacific region accounting for much of this increased risk. OCR at doses of 200 mg and 500 mg significantly inhibited the progression of structural damage and improved signs and symptoms of RA compared with MTX alone.
Acknowledgments
Support for third-party writing assistance for this manuscript was provided by F. Hoffmann-La Roche Ltd. The authors would also like to thank the FILM study investigators.
References
Footnotes
Funding F Hoffmann-La Roche and NIH grant M01-RR-00043 to the General Clinical Research Center at the University of Southern California Keck School of Medicine.
Competing interests WS has received research grants from Xencor and is a site investigator for clinical trials in RA and systemic lupus erythematosus sponsored by Genentech/Roche, Pfizer and Human Genome Sciences. JG-R has served on advisory boards for Roche, and has received consultancy fees, grants and payment for lectures from Roche. EO has received grants, consultancy fees and payments for lectures from Genentech, and consultancy fees and grants from Roche. JD has served on advisory boards for Roche Switzerland, Merck Sharp Dohme Switzerland, Pfizer Switzerland, Amgen Switzerland, UCB Switzerland, Abbott Switzerland and Novartis Switzerland, has received consultancy fees from Roche Switzerland, Merck Sharp Dohme Switzerland and UCB Switzerland, has received payment for lectures from Roche, Pfizer, Merck Sharp Dohme, Abbott and UCB, has received payment for manuscript preparation from Roche Switzerland and Merck Sharp Dohme Switzerland, and has received travel/accommodation/meeting expenses from Roche Switzerland, Merck Sharp Dohme Switzerland, Bristol Myers Squibb Switzerland and Pfizer Switzerland. RMF has received research grants from Genentech and Roche. CZ has received consultancy fees and grants from Sanofi, Servier and Pfizer, and research grants from Lilly, Amgen, Roche and Merck. AA was an employee of Genentech and held Genentech stock options during the conduct of the study and manuscript preparation. SG is an employee of Genentech, owns Roche stock and holds Roche stock options. RB and JG are employees of Roche. SF is an employee of Genentech. WD is an employee of Genentech, owns Roche stock and holds Roche stock options.
Patient consent Obtained.
Ethics approval Ethical approval was obtained from the local institutional review board at each study centre.
Provenance and peer review Not commissioned; externally peer reviewed.