Article Text

Abstract
Objective To assess the safety and effectiveness of imatinib mesylate in the treatment of diffuse cutaneous systemic sclerosis (dcSSc).
Methods In this phase IIa, open-label, single-arm clinical trial, 30 patients with dcSSc were treated with imatinib 400 mg daily. Patients were monitored monthly for safety assessments. Modified Rodnan skin scores (MRSS) were assessed every 3 months. Pulmonary function testing, chest radiography, echocardiography and skin biopsies were performed at baseline and after 12 months of treatment.
Results Twenty-four patients completed 12 months of therapy. 171 adverse events (AE) with possible relation to imatinib were identified; 97.6% were grade 1 or 2. Twenty-four serious AE were identified, two of which were attributed to study medication. MRSS decreased by 6.6 points or 22.4% at 12 months (p=0.001). This change was evident starting at the 6-month time point (Δ=−4.5; p<0.001) and was seen in patients with both early and late-stage disease. Forced vital capacity (FVC) improved by 6.4% predicted (p=0.008), and the diffusion capacity remained stable. The improvement in FVC was significantly greater in patients without interstitial lung disease. Health-related quality of life measures improved or remained stable. Blinded dermatopathological analysis confirmed a significant decrease in skin thickness and improvement in skin morphology.
Conclusions Treatment with imatinib was tolerated by most patients in this cohort. Although AE were common, most were mild to moderate. In this open-label experience, improvements in skin thickening and FVC were observed. Further investigation of tyrosine kinase inhibition for dcSSc in a double-blind randomised placebo controlled trial is warranted.
This paper is freely available online under the BMJ Journals unlocked scheme, see http://ard.bmj.com/info/unlocked.dtl
Statistics from Altmetric.com
Systemic sclerosis (SSc) is a multisystem, fibrosing disorder in which vasculopathy, autoimmunity and inflammation lead to morbidity and increased mortality.1 Abnormal signalling through the transforming growth factor beta (TGFβ)2,–,4 and the platelet-derived growth factor5,–,8 axes contributes to this pathological fibrosis. Imatinib mesylate (Gleevec; Novartis Pharmaceuticals, Basel, Switzerland) is a tyrosine kinase inhibitor with activity against c-Abl, the platelet-derived growth factor receptor (PDGFR), and other tyrosine kinases and is a therapy of interest for SSc because of its ability to interfere with both TGFβ and platelet-derived growth factor signalling.9 10
Combined inhibition of both TGFβ and PDGFR signalling by imatinib has been shown to decrease the production of extracellular matrix proteins by both scleroderma and normal skin fibroblasts.11 In murine models of both early and established cutaneous fibrosis and pulmonary fibrosis, imatinib treatment was beneficial.11,–,14 Similar effects were seen with nilotinib and dasatinib, other tyrosine kinase inhibitors that also block c-abl and PDGFR.15
Case reports of the use of imatinib in patients with SSc16,–,18 and mixed connective tissue disease19 have suggested clinical benefit. In case series of other cutaneous fibrosing disorders, nephrogenic systemic fibrosis20 and chronic sclerodermatous graft versus host disease,21 22 imatinib also showed a potential antifibrotic effect on the skin. However, in a randomised, placebo controlled study of imatinib for idiopathic pulmonary fibrosis treatment was not beneficial.23
We report here the results of a single-centre, phase IIa, single-arm, open-label clinical trial assessing the safety and efficacy of imatinib in the treatment of diffuse cutaneous systemic sclerosis (dcSSc).
Patients and methods
Study subjects
Between September 2007 and March 2009, 30 patients were enrolled. All patients fulfilled the American College of Rheumatology classification criteria for SSc24 and had the diffuse subtype. Patients were stratified by disease duration defined as onset of first symptom of SSc apart from Raynaud's phenomenon: group 1 (n=20) less than 4 years and group 2 (n=10) 4–10 years.
All subjects were over 18 years old, had a stable modified Rodnan skin score (MRSS) of 16 points or more in the month between screening and baseline visits, and had disease duration of less than 10 years. Exclusion criteria included treatment with immunosuppressive therapies 3 months before baseline (including prednisone equivalent >10 mg), pregnancy, serious medical conditions, diffusion capacity of carbon monoxide (DLCO) less than 30% predicted, or ejection fraction (EF) less than 50% (see supplementary material, available online only).
Study design
This was an investigator-initiated phase IIa, single-centre, single-arm, open-label clinical trial. The primary objective of this study was to assess the safety and tolerability of imatinib in patients with dcSSc as assessed by the number of adverse events (AE) and serious adverse events (SAE). The primary efficacy endpoint was change in the MRSS after 12 months of treatment. Secondary efficacy endpoints included change in forced vital capacity (FVC) and DLCO on pulmonary function testing (PFT) after 1 year of treatment, change in the short form 36 mental and physical components and change in scleroderma health assessment questionnaire disability index.
The protocol was approved by the institutional review board at the Hospital for Special Surgery and was conducted in accordance with the Declaration of Helsinki and good clinical practice guidelines. Patients provided written informed consent before enrollment. An independent data and safety monitoring board regularly reviewed safety data. The trial was registered at ClinicalTrials.gov (NCT00555581).
Clinical outcomes
Patients were assessed at monthly visits for AE ascertainment, interval history, physical examination and clinical laboratories, and were to call our centre for issues between visits. AE were listed according to the National Cancer Institute's common terminology.25 The MRSS was measured at screening, 1 month later at baseline and every 3 months. MRSS measurements were performed by the same physician at each visit (RS or both RS and JKG) PFT with measurement of FVC and DLCO were performed at baseline and following 12 months of therapy and are available on 22 completers.
Treatment
All patients were treated with imatinib at a target dose of 400 mg daily by mouth for 12 months. Dose modifications and interruptions were made for AE and were recorded. After 12 months of treatment, imatinib was stopped for 3 months. Patients were reassessed and offered entrance to an extension phase of the trial.
Dermatopathology
Four-millimetre punch biopsies of lesional forearm skin were performed before and after 12 months of treatment in 24 completers. The post-treatment biopsy was taken 1 cm adjacent to the original biopsy. Nineteen pairs were bisected. Half were formalin-fixed and paraffin-embedded, and half were stored for RNA extraction. The five final samples were prospectively reserved and submitted in entirety for RNA extraction to preserve adequate RNA quantity for microarray and follow-up confirmatory studies. The six patients who did not complete the study did not have follow-up biopsies. Sections were stained with H&E, anti-α-smooth muscle actin, Masson trichrome and elastin using standard techniques. A dermatopathologist (CM), blinded to before or after treatment status, compared the paired specimens.
Statistical analysis
The primary endpoint was a description of AE and SAE, and for this descriptive data are provided. No power calculation was performed for safety measurements, because there was no statistical analysis to be performed without a comparison group. The MRSS was the primary efficacy endpoint. Given a 5±8 unit difference in MRSS, based on a minimally clinically important difference of 5 points,26 a sample size of 30 was needed for a two-sided alpha of 0.05 and 90% power. Analysis of all patients remaining on medication and reaching efficacy endpoints at months 3 (n=27), 6 (n=26), 9 (n=26) and 12 (n=24) is shown below as per our predefined protocol. A post-hoc modified intent-to-treat analysis including all patients with follow-up data, was also performed and was not different (not shown). In the case of missing data points, we utilised last observation carried forward. Paired t tests and Wilcoxon signed-rank tests were used to compare outcome measures before and after treatment, and unpaired t tests were used for comparisons between groups of patients. Statistical analysis was performed using SPSS software version 17.0.
Results
Fifty-three patients were screened, 35 met entry criteria and five declined consent. Thirty patients started medication and 24 completed the study protocol. Baseline characteristics of the 30 subjects are summarised in table 1. Patients were categorised as having interstitial lung disease (ILD) if they had evidence of ground glass opacity and/or fibrosis on CT of the chest felt to be related to SSc in the opinion of the investigators and the treating physicians..
Baseline demographics, scleroderma related organ system involvement and previous treatments
Three patients withdrew from the study in the first 3 months: two because of non-compliance and one developed an inflammatory myopathy that required immunosuppression. One patient withdrew after 3 months because of myalgia and fatigue, and one patient withdrew after 11 months because of the development of multiple medical issues (described below). One patient died after 11 months on medication. Safety data are included on all 30 patients enrolled.
Safety outcomes
Adverse events
There were 358 total AE captured during the study period, of which 171 were considered to be possibly, probably, or definitely related to imatinib. All AE at least possibly related to imatinib and occurring in more than one patient are presented in table 2. Of these 171 AE, 72.5% were grade 1, 25.1% were grade 2 and 2.4% were grade 3 or 4 in severity. The most common AE was oedema, which was seen in 80% of patients. Fluid-related issues required the addition of furosemide in 60% of patients and dose adjustment in 30%. All AE captured regardless of attribution are presented as supplementary material, available online only.
AE at least possibly related to imatinib, ocurring in more than one patient and all SAE
Eighty-three per cent of patients required dose adjustment at least one time, and the median dose taken as assessed by both pill count and patient report was 300 mg daily. The leading causes for dose adjustment or interruption were musculoskeletal complaints in 36.7%, fluid-related issues in 26.7%, intercurrent illness in 23.3%, gastrointestinal complaints in 23.3% and constitutional symptoms in 20% of patients.
Creatine kinase (CK) elevations greater than 1.5 times the baseline CK value without change in strength were seen in 43% of patients. Elevation of CK has been recognised in patients with chronic myeloid leukaemia (CML) and gastrointestinal stromal tumour with imatinib therapy.27 28 One patient, who had discontinued methotrexate 3 months before study entry, experienced increased proximal muscle weakness accompanied by an increased CK from 320 to 468 after 4 weeks on protocol. Electromyography showed inflammatory myopathy, and the patient was withdrawn from the study to be treated with immunosuppressive agents.
Serious adverse events
There were 24 SAE (table 2). One patient with severe ILD and pulmonary artery hypertension died from pneumonia and respiratory failure. As the patient was not neutropenic, had tolerated imatinib without evidence of fluid overload for 11 months, and there was no apparent volume overload during the event; the death was not felt to be medication related. Thirteen of the 24 SAE, including all seven hospitalisations for haemorrhagic cystitis, occurred in a single patient and were primarily complications of previous treatment with high-dose cyclophosphamide with autologous stem cell transplant. The haemorrhagic cystitis was a pre-existing problem before trial entry. One imatinib-related SAE was fluid overload with bilateral pleural effusions, which required hospitalisation for diuresis. This episode occurred in the context of the patient receiving fluid resuscitation for a severe haemorrhoidal bleed. The patient subsequently tolerated reintroduction and treatment with imatinib 300 mg daily with concomitant furosemide administration.
Chest radiography and echocardiography
Postero-antero and lateral chest radiographs and transthoracic two-dimensional echocardiograms were performed at baseline and after 12 months of treatment. Twenty-three of 24 chest radiographs were unchanged. One patient with ILD developed increased infiltrates on the follow-up study, which was further evaluated with high resolution CT and found to be unchanged from baseline.
Three patients had echocardiographic changes during the course of the trial, two of which were of clinical significance. One patient, who did not complete the trial, developed cardiomyopathy with congestive heart failure at a point when she had largely discontinued the medication. The patient's treating physicians felt the cardiomyopathy was a result of ischaemic heart disease and was tachyarrhythmia mediated, and the investigators attributed this SAE as possibly related to imatinib. Two additional patients had declines in EF as noted on surveillance echocardiography from baseline normal EF to EF of 50–55%. One patient followed up with a cardiac MRI and was found to have a normal EF of 60%. The other patient underwent a multigated image acquisition scan, which confirmed the decline in EF. This patient had experienced disease progression in pulmonary, gastrointestinal and cutaneous manifestations, and the cardiomyopathy was felt to be most likely related to the progression of disease.
Modified Rodnan skin score
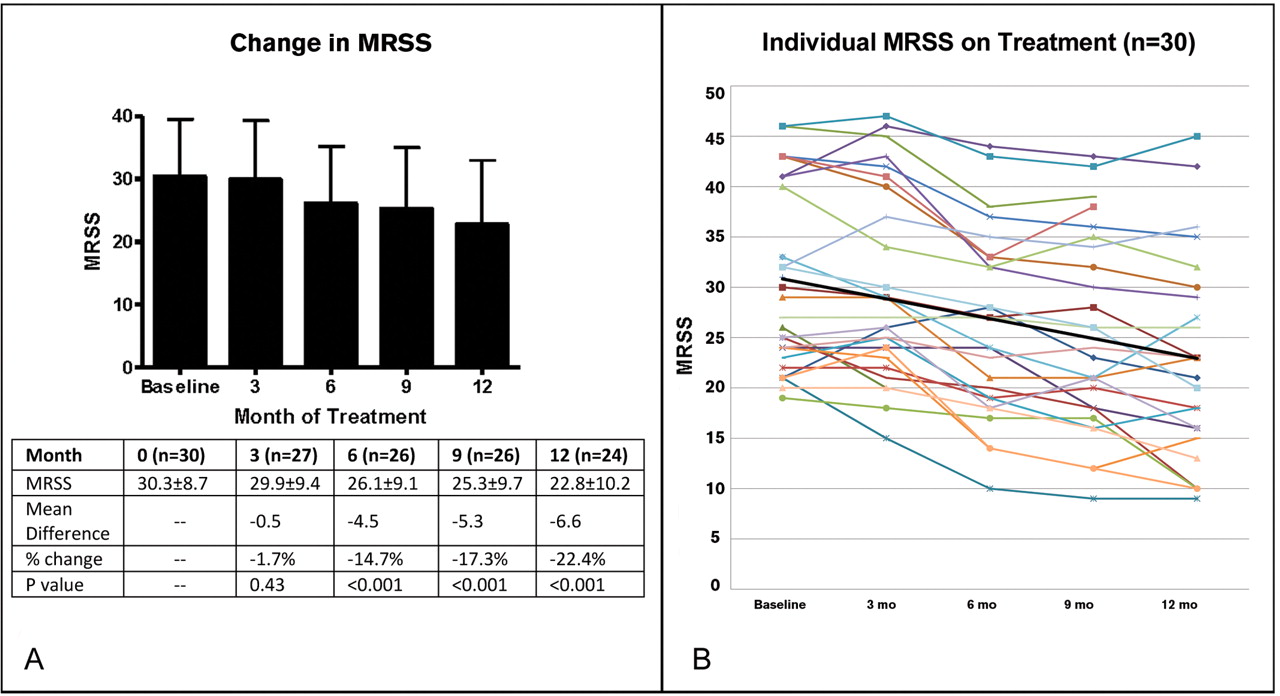
There was a significant improvement of the MRSS over the course of 1 year with treatment with imatinib. At 12 months of treatment, the mean MRSS improved from 29.42±8.6 to 22.8±10.2 (p<0.001) and the mean difference in MRSS was −6.6 points (95% CI −4.5 to −8.7). This change was not seen after 3 months of treatment, but was evident at 6 months, with a mean improvement of −4.5 points (−2.6 to −6.5; p<0.001) and at 9 months with a mean improvement of −5.3 points (−3.5 to −7.1; p<0.001; figure 1).
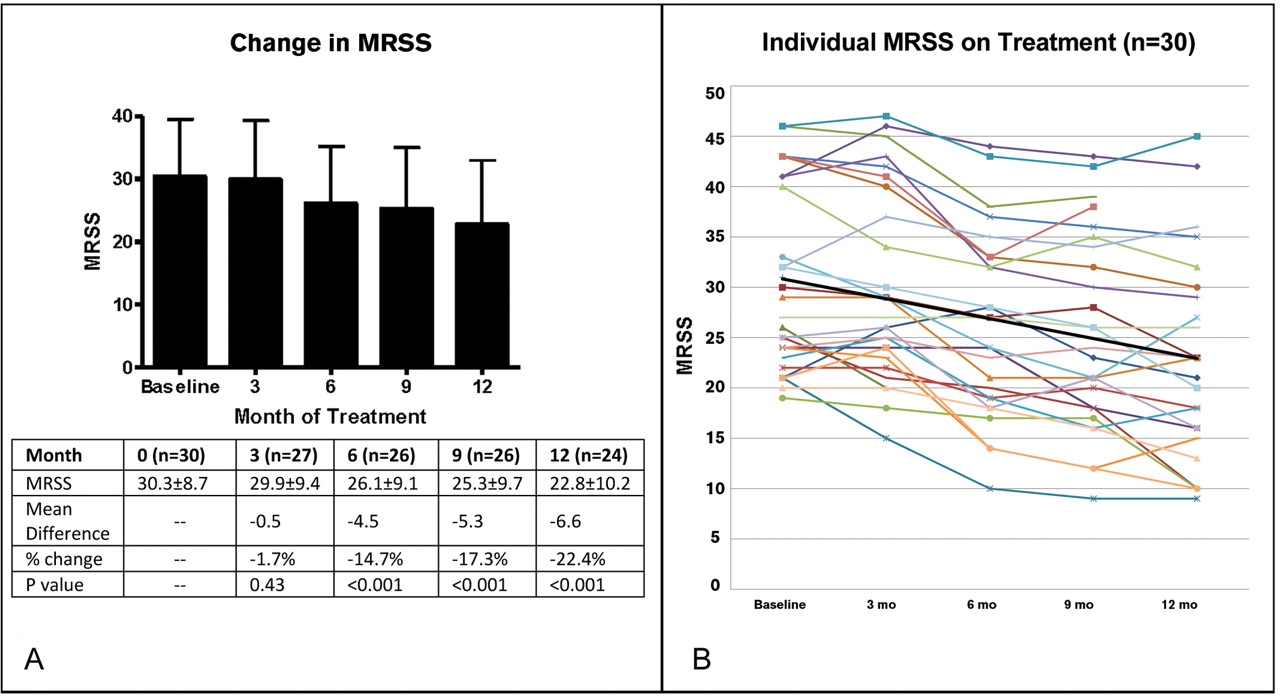
Modified Rodnan skin score (MRSS) over the duration of the trial in all patients on treatment. At baseline the MRSS was 30.3±8.7 (n=30). After 3 months of imatinib therapy the MRSS was 29.9±9.4 compared with a baseline mean of 30.4±9.1 in this group (n=27); p=0.428. After 6 months the MRSS was 26.1±9.1; p<0.001 compared with baseline mean of 30.6±9.2 in this group (n=26). After 9 months the MRSS was 25.3±9.7; p<0.001 (n=26). After 12 months of treatment the mean MRSS was 22.8±10.2 compared with a baseline MRSS of 29.4±8.6 in this group (n=24); p<0.001. (A) As bar chart and (B) as individual patient plots. Black line is mean trendline.
Similar improvements in MRSS were seen across different durations of disease. In a post hoc analysis of patients with early disease of less than 18 months' duration (n=8), there was a significant improvement of 7.9±5.7 points (p=0.006; table 3). There were no differences in change in MRSS between disease duration subgroups whether 18 months or 4 years was used as the cut-off for defining ‘early’ disease. There was no difference in change in MRSS between patients who were anti-Scl70 positive versus those who were anti-Sc170 negative.
Change in MRSS by disease duration
Pulmonary function testing
FVC improved from a mean of 82.9±21.1% to 89.3±25.2% predicted (p=0.008). The mean DLCO trended from 78.0±22.9 % to 83.5±29.2% predicted (p=0.12; figure 2A,B). When patients were subcategorised by the presence or absence of ILD, those with ILD remained stable with respect to their FVC and DLCO, with an average increase in FVC of 2.1% points (95% CI −2.9 to 7.0; p=0.36) and an average increase in DLCO of 1.0% points (−8.0 to 10.1; p=0.81). Those without ILD improved with respect to their FVC, with an average increase of 10.7% points (−3.3 to 18.2; p=0.01), and remained stable with respect to their DLCO, with an average change of +9.9% points (−1.7 to 21.5; p=0.09). Patients without ILD had a greater improvement in their change in FVC than did those with ILD (p=0.043) but there was no difference between the groups with respect to the change in DLCO (p=0.19; table 4). Differences were not seen in PFT changes based on disease duration or based on anti-scl70 status.
Outcome measures
Additional outcomes
Outcomes regarding health-related quality of life measures are shown in table 4, with significant improvements or stability in multiple parameters. There was no correlation between change in FVC and change in MRSS (Spearman's rank correlation −0.071).
Dermatopathology
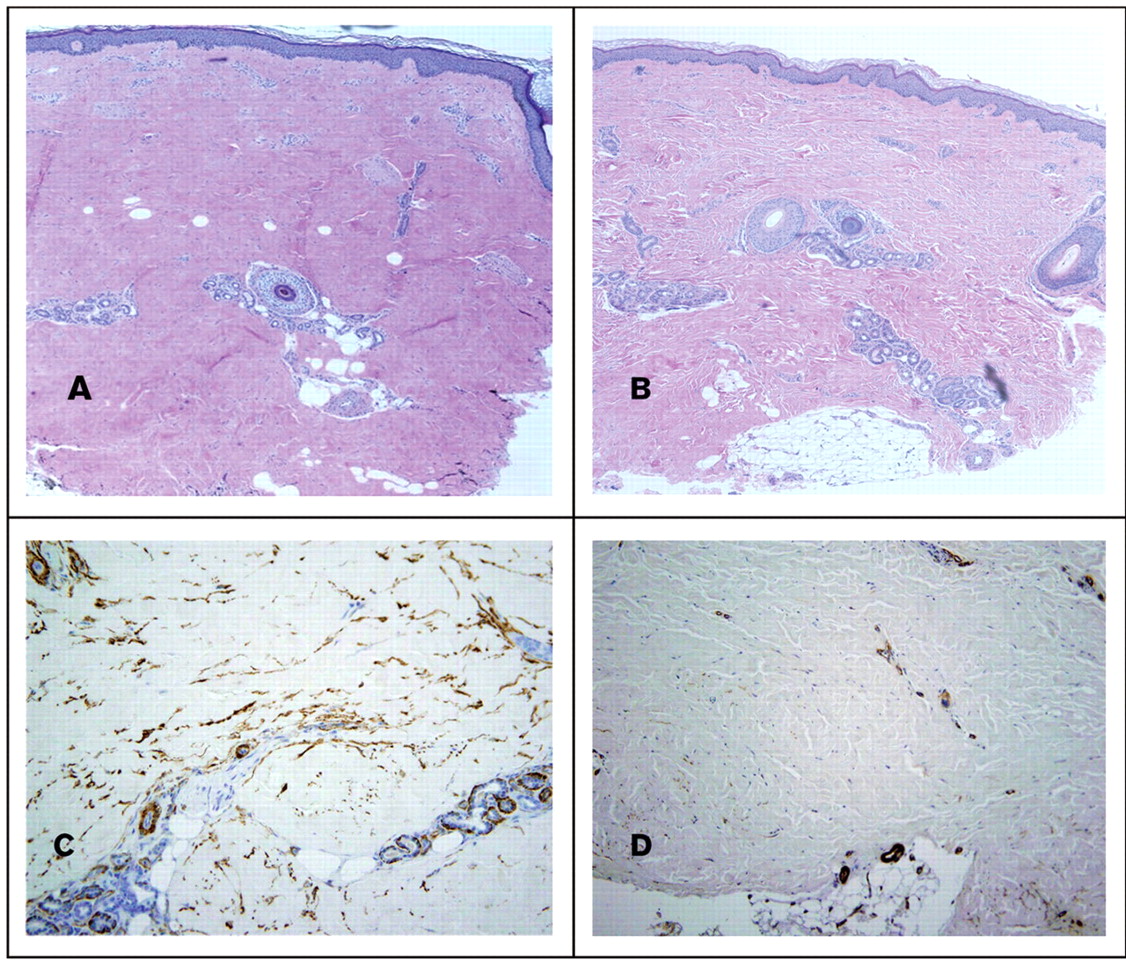
The median skin thickness decreased with treatment from 2.23 mm (IQR 2.0–2.95) to 2.0 mm (1.81–2.36) as measured from the granular cell layer to the dermal subcutaneous interface or the greatest depth on H&E (p<0.01). There was a significant increase in the number of appendageal structures (hair follicles plus eccrine glands) counted per vertical section from two (one to four) to three (two to six); p=0.03. All specimens before treatment exhibited changes typical of scleroderma including thick and hyalinised collagen bundles with decreased interstitial spaces. Nine of the pretreatment specimens exhibited artefact from overfixation, which precluded comparative assessment with the post-treatment specimen. Of the 10 assessable specimens, seven exhibited a qualitative decrease in the thickness of collagen bundles and increase in interstitial spaces (figure 2). One specimen had increased sclerodermatous characteristics and two were unchanged. The morphological differences corresponded to MRSS improvement with the mean change in MRSS in the group with histological improvement being −8.8 (−6 to −12), the unchanged biopsies had MRSS changes of −1 and −5, and the patient whose biopsy looked worse had an MRSS of +4. Eight before and after pairs stained adequately with anti-α-smooth muscle actin. Of these, three showed decreased fibrocyte staining after treatment, one showed increased staining and four were unchanged. No differences were discernible with elastin or Masson trichrome staining.
{kind=link}
{kind=link}
(A–D) Depicted are skin biopsy specimens before and after 12 months of imatinib therapy in a single patient at 4× magnification. (A and B). H&E: After treatment there was a decrease in skin thickness. In the post-treatment specimen the collagen bundles are less thick and there is an increase in the interstitial spaces between the bundles. There are also increased numbers of adnexal structures in the post-treatment specimen. This individual patient is anti-Scl70 positive, with a disease duration of 4 months at baseline who had an improvement in MRSS of 9 points over the course of 12 months. In C and D are depicted anti-α-smooth muscle actin staining before treatment in panel C and post-treatment in D, showing a decline in the intensity of staining.
Discussion and conclusions
Tyrosine kinase inhibition has emerged as a therapeutic approach of interest in scleroderma and other fibrosing disorders. This study represents the largest prospective trial of imatinib in dcSSc reported to date. Our results indicate acceptable safety and tolerability, and suggest the potential efficacy of imatinib for cutaneous and pulmonary manifestations of dcSSc, as well as benefit in patient-derived subjective outcomes. The strengths of this study include the number of patients treated, its prospective design and its relatively long duration. The obvious weakness is the open-label design and lack of a control group, making attribution of improvements in clinical outcomes as well as of AE and SAE uncertain. Furthermore, this was a heterogeneous population in terms of disease duration, organ system involvement and previous treatment history, making it difficult to compare this experience with trials including only early active SSc patients.
While patients in our trial had relatively early disease (mean disease duration 3.4 years), we prospectively elected to enroll 10 patients with later disease (4–10 years), appreciating that this subgroup is also in need of better therapies. Their inclusion in this pilot study was necessary to establish the tolerability and safety of this potentially antifibrotic therapy in later-stage patients, which would be requisite for their inclusion in future controlled studies. We recognise that including this subgroup adds to the difficulty in interpreting the significance of the improvements observed in our trial. Encouragingly, however, similar improvements were also seen in patients with very early (<18 months) disease duration. Nevertheless, definitive conclusions regarding efficacy cannot be drawn from an open-label experience, especially noting that spontaneous improvements in skin scores can be seen even in patients with early stage dcSSc in clinical trials29 or observational studies.30 Although the blinded dermatopathological assessments support our clinical findings, attribution of these improvements to treatment is uncertain without a longitudinal control group.
A large number of AE and SAE were recognised during this 1-year trial, which may relate in part to our patients' significant disease burden. However, 80% of patients enrolled were able to complete a full year of therapy. This number compares favourably to what has been observed in other clinical trials in scleroderma,31,–,34 but not surprisingly is less than what has been seen in trials of CML.35 Oedema, myalgia and fatigue were frequent side effects, but were manageable, and infrequently led to withdrawal from our study. Cardiac toxicity has been reported as a rare complication of imatinib in CML, particularly in patients with concurrent risk factors for cardiomyopathy,36 making it an important concern in SSc. Two patients in our study did develop cardiac issues that were not felt to be likely related to imatinib given the individuals' clinical contexts; nevertheless, vigilance for this potential complication is prudent.
Our recognition of improvement in indices of pulmonary function during this 1-year trial is of unclear significance. There was a statistically significant improvement in FVC and stabilisation of DLCO at 1 year. However, not all of our patients had ILD, and the clinical significance of this improvement37 in patients without appreciable ILD at baseline is uncertain. One possible explanation is that this observed improvement in FVC may relate to amelioration of chest wall tightening rather than effects on parenchymal lung disease. Importantly, a randomised double-blind placebo controlled trial in idiopathic pulmonary fibrosis did not show benefit of imatinib therapy.36
In conclusion, this study represents the largest and longest open-label trial of imatinib in SSc. Side effects were common, but most patients tolerated the medication. Signals of potential efficacy were observed in MRSS, FVC and patient-derived quality of life indices. These findings cannot be definitively attributed to medication effect in an uncontrolled trial but deserve further investigation. A prospective randomised, double-blind, placebo controlled trial is warranted to define more clearly whether there is a role for tyrosine kinase inhibition in the treatment of dcSSc.
References
Supplementary materials
Web Only Data
Files in this Data Supplement:
Footnotes
-
Funding This study was supported by the Rudolph Rupert Scleroderma Program at the Hospital for Special Surgery and by Novartis Pharmaceuticals who provided drug as well as partial financial support. JKG was supported by an Arthritis Foundation New York Chapter Fellowship and by grant UL1RR024996 of the Clinical and Translational Science Center at Weill–Cornell Medical Center.
-
Competing interests None.
-
Ethics approval This study was conducted with the approval of the Hospital for Special Surgery and Weill–Cornell Medical Center.
-
Patient consent Obtained.
-
Provenance and peer review Not commissioned; externally peer reviewed.