Article Text

Abstract
Objective Evaluate apremilast efficacy across various psoriatic arthritis (PsA) manifestations beginning at week 2 in biological-naïve patients with PsA.
Methods Patients were randomised (1:1) to apremilast 30 mg twice daily or placebo. At week 16, patients whose swollen and tender joint counts had not improved by ≥10% were eligible for early escape. At week 24, all patients received apremilast through week 52.
Results Among 219 randomised patients (apremilast: n=110; placebo: n=109), a significantly greater American College of Rheumatology 20 response at week 16 (primary outcome) was observed with apremilast versus placebo (38.2% (42/110) vs 20.2% (22/109); P=0.004); response rates at week 2 (first assessment) were 16.4% (18/110) versus 6.4% (7/109) (P=0.025). Improvements in other efficacy outcomes, including 28-joint count Disease Activity Score (DAS-28) using C reactive protein (CRP), swollen joint count, Health Assessment Questionnaire-Disability Index (HAQ-DI), enthesitis and morning stiffness severity, were observed with apremilast at week 2. At week 16, apremilast significantly reduced PsA disease activity versus placebo, with changes in DAS-28 (CRP) (P<0.0001), HAQ-DI (P=0.023) and Gladman Enthesitis Index (P=0.001). Improvements were maintained with continued treatment through week 52. Over 52 weeks, apremilast’s safety profile was consistent with prior phase 3 studies in psoriasis and PsA. During weeks 0–24, the incidence of protocol-defined diarrhoea was 11.0% (apremilast) and 8.3% (placebo); serious adverse event rates were 2.8% (apremilast) and 4.6% (placebo).
Conclusions In biological-naïve patients with PsA, onset of effect with apremilast was observed at week 2 and continued through week 52. The safety profile was consistent with previous reports.
Trial registration number NCT01925768; Results.
- spondyloarthritis
- Das28
- disease activity
- treatment
- psoriatic arthritis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Psoriatic arthritis (PsA) is heterogeneous, with patients exhibiting varied clinical symptoms, severity and disease course. Treatment goals include controlling disease activity, optimising functional status and minimising side effects to therapy.1 Biologicals are commonly used after or in conjunction with conventional synthetic disease-modifying antirheumatic drugs (csDMARDs), but safety monitoring and risks may limit their long-term use.2 3
The efficacy and safety of apremilast, an oral phosphodiesterase 4 inhibitor, were demonstrated in patients with active PsA in four phase III, placebo-controlled studies as part of the Psoriatic Arthritis Long-term Assessment of Clinical Efficacy (PALACE) clinical trial programme.4–7 The PALACE 1, 2 and 3 studies evaluated apremilast in patients with prior exposure to csDMARDs and/or biologicals and allowed concomitant csDMARD use.4–6 PALACE 4 evaluated apremilast monotherapy in csDMARD-naïve and biological-naïve populations.7 Data demonstrating apremilast’s efficacy across disease manifestations have been reported at week 164–6 8 and up to 4 years of treatment.9 However, time to onset of therapeutic effect has not been reported before week 16.
Assessing Apremilast Monotherapy in a Clinical Trial of BIologic-NaïVE Patients With Psoriatic Arthritis (ACTIVE) aimed to evaluate apremilast monotherapy in biological-naïve PsA patients who may have had one prior csDMARD. ACTIVE also aimed to determine the onset of apremilast efficacy, with assessments beginning at week 2, and to examine additional outcome measures, including morning stiffness and enthesitis using the Gladman Enthesitis Index (GEI).10 Diarrhoea adverse events (AEs) were further characterised using a protocol definition.
This report describes the early onset and overall efficacy and safety of apremilast monotherapy through week 52.
Methods
Patients
Enrolled adults (≥18 years of age) had a documented diagnosis of active PsA for ≥3 months and met Classification Criteria for Psoriatic Arthritis.11 At screening, patients were required to have at least three swollen and three tender joints, C reactive protein (CRP) of ≥0.2 mg/dL and be biological DMARD-naïve. No csDMARD washout before the study was required (except 4 weeks for cyclosporine and 12 weeks for leflunomide); however, patients had to discontinue their current csDMARD ≥1 day before baseline assessments. Patients were excluded if they had prior treatment with more than one csDMARD; used prohibited systemic therapies, including cyclosporine or other calcineurin inhibitors, within 4 weeks of randomisation, corticosteroids >10 mg daily (prednisone or equivalent), oral agents such as retinoids, mycophenolate, thioguanine, hydroxyurea, sirolimus and tacrolimus; and inflammatory joint disease other than PsA. Also excluded were patients with active or incompletely treated tuberculosis, significant infection within 4 weeks of screening and current or history of malignancy (except for treated basal cell or squamous cell skin carcinoma or early forms of cervical carcinoma with no recurrence within 5 years).
All patients provided written informed consent before any study procedures were initiated.
Study design
This phase IIIB, multicentre, randomised, double-blind, placebo-controlled, parallel-group study evaluated the efficacy and safety of apremilast monotherapy in patients with active PsA.
Patients were randomised (1:1) to apremilast 30 mg twice daily or placebo for 24 weeks, stratified by previous csDMARD and baseline prednisone (or equivalent) use. Patients who did not improve by ≥10% in swollen joint count (SJC) and tender joint count (TJC) at week 16 were eligible for early escape at the investigator’s discretion. Early escape patients initially randomised to placebo were switched to apremilast in blinded fashion, with dose titration during the first week of treatment; patients initially randomised to apremilast remained on apremilast. At week 24, all remaining patients receiving placebo switched to apremilast for the active treatment phase through week 52, when all patients were eligible to continue apremilast treatment in an open-label extension phase through week 104.
Concomitant medications
Patients could receive concurrent treatment with stable doses of oral corticosteroids (prednisone ≤10 mg/day or equivalent), non-steroidal anti-inflammatory drugs or opioid analgesics. Changes in corticosteroid doses and/or discontinuations were not allowed from day 0 to week 24 except for safety reasons or lack of availability. After week 24, the corticosteroid dose could be adjusted as clinically required. Patients could use low-potency topical corticosteroids for face, axillae and groin psoriatic lesions.
Outcomes
The primary outcome was 20% improvement in modified American College of Rheumatology response criteria (ACR20) at week 16. Other efficacy outcomes included 28-joint count Disease Activity Score (DAS-28) using CRP, SJC, TJC, six-point GEI score (0=no enthesitis, 6=all six sites active (ie, bilateral tibial tuberosity, plantar fascia and Achilles tendon insertion)) for patients with enthesitis at baseline, morning stiffness duration and severity, ACR50 and ACR70 and physical function assessments, including the Health Assessment Questionnaire-Disability Index (HAQ-DI), 36-item Short-Form Health Survey version 2 (SF-36v2) Physical Functioning (PF) domain and physical component summary (PCS) scores. Safety and tolerability evaluations included collection of AEs, vital signs, laboratory evaluations, physical examinations, electrocardiograms, chest X-rays and further characterisation of diarrhoea AEs using a protocol definition (two or more watery or liquid stools/day).
Efficacy outcomes were assessed starting at week 2 and at scheduled visits through week 52; SF-36v2 assessments started at week 4.
Statistical analysis
Efficacy analyses were based on the full analysis set, which included all randomised patients. The safety population included all randomised patients who received at least one dose of study medication. Sample size estimation was based on results from earlier phase III studies. A two-group χ2 (continuity-corrected) test with a two-sided 0.05 significance level would have ≈90% power to detect a true 20% difference (35% vs 15%) between apremilast and placebo for the proportion of patients achieving ACR20 response at week 16, when the sample size in each group was 107.
Baseline patient demographics and disease characteristics were compared descriptively between the treatment groups.
For the placebo-controlled period, two-sided tests for efficacy outcomes were performed sequentially according to a prespecified hierarchical order to control the overall type I error rate (online supplementary table 1). P values <0.05 were considered statistically significant; if the P value did not reach the threshold of 0.05 during the hierarchical testing, the nominal P value was reported onwards. Therefore, P values <0.05 should be interpreted with caution for the secondary outcomes if a testing in a higher order of the hierarchy did not reach the threshold of 0.05.
Supplementary file 1
Dichotomous variables such as ACR20 response were analysed using the generalised Cochran-Mantel-Haenszel test,12 controlling for baseline prednisone (or equivalent) use (yes/no) and previous csDMARD use (yes/no). Patients escaping at week 16 were primarily treated as non-responders at the subsequent time points during the placebo-controlled period. Missing data were handled using non-responder imputation. Mixed-effect model for repeated measures was generally used for analyses of continuous variables such as HAQ-DI, where change or per cent change from baseline was the dependent variable and treatment group, time (ie, study week), treatment-by-time interaction, baseline prednisone (or equivalent) use (yes or no) and previous DMARD use (yes or no) were factors and baseline value was a covariate. Time was treated as a categorical variable in the mixed-effect model for repeated measures. Data obtained after early escape were excluded from the model.
Week 52 efficacy data descriptions were as-observed analyses when no placebo data were available for comparison.
Safety analyses were conducted for the placebo-controlled phase (weeks 0–24) and overall apremilast-exposure period, which includes all available safety data among patients who received at least one dose of apremilast at any time up to the data cut-off, at which time all patients remaining in the study had completed their week 52 visit. AEs were classified using MedDRA V.14.0.
Results
Patients
A total of 219 patients were randomised (apremilast: n=110; placebo: n=109), and 84.5% completed week 24 (online supplementary figure 1). Of the 180 patients entering the long-term active treatment phase, 88.9% completed week 52. Treatment groups were comparable for baseline patient demographics and disease characteristics (table 1).
Baseline patient characteristics (full analysis set)
Efficacy
Primary outcome: ACR20 response
The ACR20 response rate at week 16 was significantly greater in patients receiving apremilast versus placebo (38.2% (42/110) vs 20.2% (22/109); P=0.004) (table 2), with response observed at week 2 (16.4% (18/110) vs 6.4% (7/109)); P=0.025). At week 24, greater improvements in ACR20 response rate were observed with apremilast versus placebo (43.6% (48/110) vs 24.8% (27/109); P=0.004).
Efficacy outcome measures at week 2, week 16 and week 52†
Evidence of treatment effect in various additional PsA manifestations was observed with apremilast at week 2 (first evaluation after baseline), as assessed by DAS-28 (CRP), HAQ-DI, GEI and morning stiffness severity (table 2).
Disease activity, joint count, enthesitis and morning stiffness outcomes
Efficacy was seen across a number of secondary measures assessing disease activity, joint inflammation, enthesitis and morning stiffness (table 2). At week 16, apremilast-treated patients demonstrated a significant reduction from baseline in DAS-28 (CRP) score versus placebo (P<0.0001) (table 2). Reductions continued through week 24 (−1.26 vs −0.76; P=0.005).
Significant improvement was demonstrated by mean per cent change in SJC with apremilast versus placebo at week 16 (P=0.0001) (table 2), with continued improvements detected at week 24 (−59.1% vs −29.0%; P=0.002). Mean per cent changes in TJC were significant with apremilast versus placebo at week 16 (P=0.002) (table 2) and week 24 (−49.6% vs −25.3%; P=0.009).
Among patients with enthesopathy at baseline (apremilast: n=56; placebo: n=51), significant improvements in enthesitis counts were observed at week 16 (P=0.001) with apremilast versus placebo. Improvements were observed at week 2 (P=0.035) and continued to week 24 (−1.5 vs −0.5; P=0.003). Numerically greater proportions of apremilast patients achieved a GEI score of 0 through week 24 (44.6% (25/56) vs 33.3% (17/51)).
Improvements in morning stiffness duration were observed with apremilast versus placebo at week 16 (P=0.005) (table 2) and week 24 (median per cent change: −33.3% vs 0.0%; P=0.001). More apremilast-treated patients showed improvement in morning stiffness severity at week 16 (P=0.015) (table 2) continuing to week 24 (40.0% vs 20.2%; P=0.002).
Functional ability
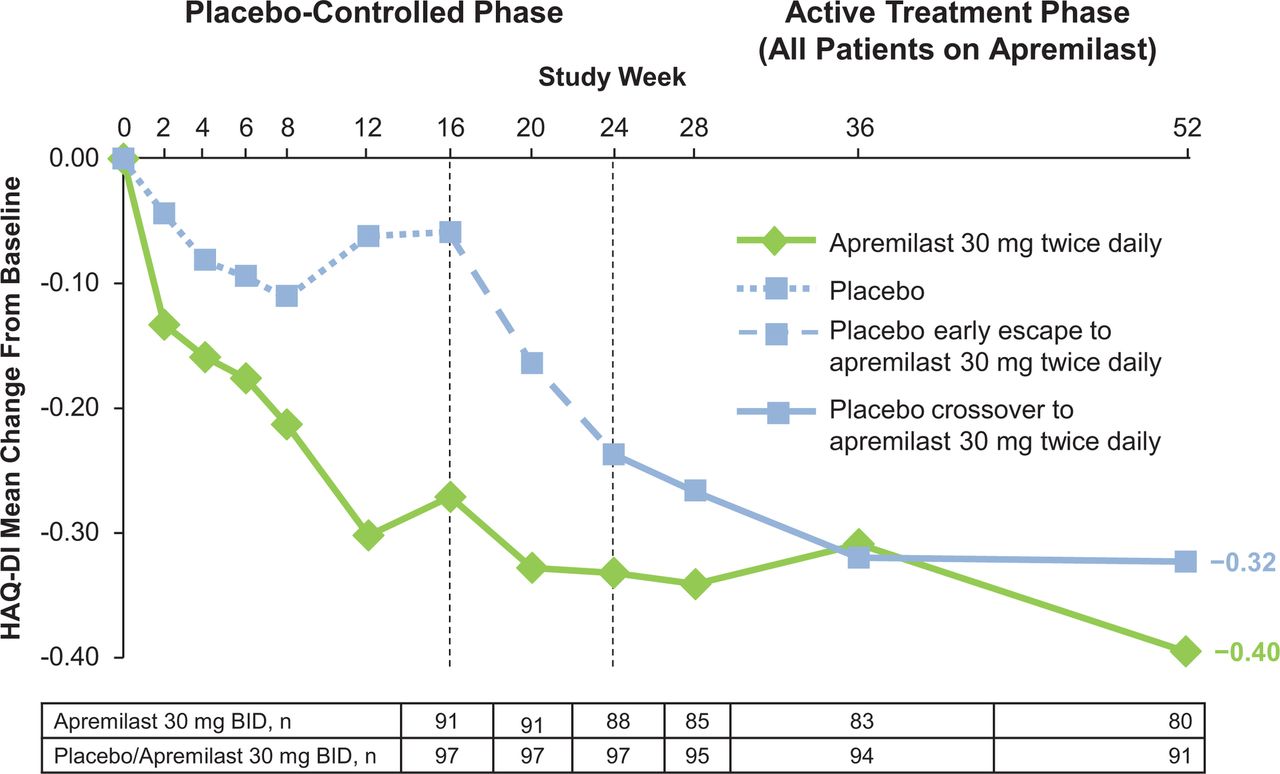
Apremilast-treated patients experienced improvements in physical disability, as assessed by various outcomes for physical function. Clinically meaningful and significant improvements were observed in physical function, as indicated by decreases in HAQ-DI score at week 16 with apremilast versus placebo (−0.21 vs −0.06; P=0.023). Decreases were observed beginning at week 2 (P=0.040) (table 2). The improvements seen with apremilast continued through week 24, with a mean reduction of −0.27; however, the mean change did not reach statistical significance versus placebo due to an unexpected shift in mean improvement in the placebo group between weeks 16 and 24 (−0.27 vs −0.17; P=0.168).
Notably, mean changes in HAQ-DI score with apremilast met or exceeded the minimal clinically important difference (MCID) of −0.13 (prespecified analysis)13 at weeks 2, 16 and 24. The proportion of patients achieving an MCID ≥0.35 (post hoc analysis)14 was numerically higher with apremilast versus placebo at week 16 (table 2) and significantly higher with apremilast versus placebo at week 24 (40.9% vs 24.8%, P=0.014).
Significant improvement in physical function was demonstrated by improvements from baseline in SF-36v2 PF score with apremilast versus placebo at week 16 (P=0.004) (table 2). Continued SF-36v2 PF improvement was observed at week 24 with apremilast versus placebo (3.94 vs 1.26; P=0.017), with least-squares mean improvement exceeding the MCID of 2.5.15 Similarly, significant improvements in the SF-36v2 PCS score were observed with apremilast versus placebo at week 16 (P=0.0001) (table 2) and at week 24 (5.00 vs 1.60; P=0.004), and the least-squares mean improvement at each time point with apremilast exceeded the MCID of 2.5.15
Subset analysis
In a subset of patients (69% of overall population) who had one prior csDMARD, significant ACR20 response rates were observed with apremilast versus placebo (39.2% (29/74) vs 20.5% (16/78); P=0.013) at week 16. These rates were similar to those observed in the overall population. Improvements in joint and enthesitis outcomes in the subset were also similar to those observed in the overall population. In the subset, the week 16 mean per cent change with apremilast versus placebo was −40.7% versus 3.1% (P=0.003) for SJC and −26.8% versus 5.4% (P=0.014) for TJC; mean change in GEI score was −1.51 versus −0.18 (P=0.001) (online supplementary table 2). Similar results were observed in the subset (58% of overall population) with prior methotrexate use.
Long-term durability
Clinical improvements across outcomes, including swollen and tender joints, enthesitis, morning stiffness and functional ability, were sustained through week 52 (table 2; figures 1–3; online supplementary figure 2); for individuals who received apremilast from baseline, mean per cent change in SJC was −77.5%, with 55.0% (44/80) achieving SJC ≤1, and mean per cent change in TJC was −70.4%, with 42.5% (34/80) achieving TJC≤1.

(A) ACR20 response, (B) mean per cent change in SJC and (C) mean per cent change in TJC through week 52. All data shown are as observed among patients as randomised at baseline and receiving at least one dose of apremilast. ACR20, 20% improvement in modified American College of Rheumatology response criteria; n/m, number of responders/number of patients with sufficient data for evaluation; SJC, swollen joint count; TJC, tender joint count.
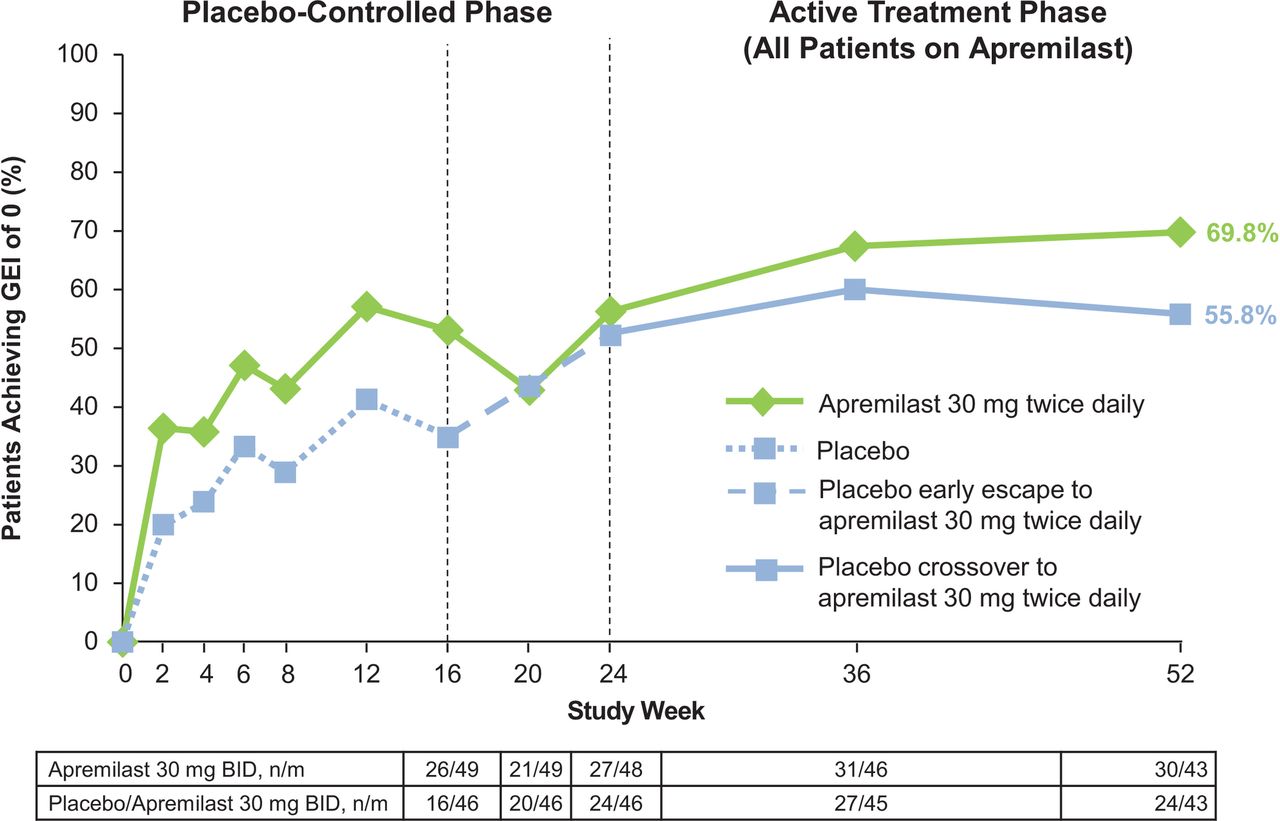
Proportion of patients achieving a GEI of 0* through week 52. All data shown are as observed among patients as randomised at baseline, receiving at least one dose of apremilast and having pre-existing enthesopathy at baseline (eg, GEI score >0, n=102). GEI, Gladman Enthesitis Index; n/m, number of responders/number of patients with sufficient data for evaluation.
{kind=link}
{kind=link}
{kind=link}
Mean change from baseline in HAQ-DI score through week 52. All data shown are as observed among patients as randomised at baseline and receiving at least one dose of apremilast. HAQ-DI, Health Assessment Questionnaire-Disability Index.
Safety
During the placebo-controlled phase (weeks 0–24), mean total exposure duration was 20.03 weeks (41.8 patient-years) for placebo patients and 20.93 weeks (43.7 patient-years) for apremilast patients. During the apremilast-exposure period, mean total duration of apremilast exposure was 52.1 weeks (205.6 patient-years).
Overall AE incidence through week 24 was generally similar between the apremilast and placebo groups (table 3). The most commonly reported AEs (≥5% of either treatment group) during the placebo-controlled phase were diarrhoea, nasopharyngitis, nausea, headache, hypertension and upper respiratory tract infection (table 3). During weeks 0–24, a total of 15 patients (apremilast: n=10; placebo: n=5) discontinued because of AEs. The nature, incidence and severity of AEs were comparable with longer apremilast exposure. Six patients (five randomised to placebo at baseline; one randomised to apremilast at baseline) discontinued after week 24 because of AEs (online supplementary figure 1).
Nature, incidence and severity of AEs
Serious AEs were low for both groups (apremilast: 2.8%; placebo: 4.6%) during the placebo-controlled phase; none were considered drug related. No serious opportunistic infections, including new or reactivated tuberculosis, were reported during the study. One death occurring after week 52 was due to atherosclerotic cardiovascular disease in a patient with a pre-existing history of hypertension and alcoholic cardiomyopathy (discovered at autopsy).
Diarrhoea was the most frequently reported AE during the placebo-controlled phase (apremilast: 14.7%; placebo: 11.0%); all cases were mild to moderate in severity. A protocol definition of diarrhoea was applied to further characterise the diarrhoea events. Using the criteria of two or more watery/liquid stools/day, 21 patients had diarrhoea (apremilast: n=12 (11.0%); placebo: n=9 (8.3%)) during the placebo-controlled phase. Four of these diarrhoea events led to study discontinuation in apremilast-treated patients. Three (apremilast: n=1; placebo: n=2) of the 21 patients took antidiarrhoeal medications. From week 24 to week 52, 10 new patients experienced protocol-defined diarrhoea AEs. Onset of diarrhoea (including protocol-defined diarrhoea AEs) was most frequently observed during the first 4 weeks of dosing. No evidence of increased gastrointestinal events was observed during the longer apremilast-exposure period versus the placebo-controlled phase.
No cases of suicidal ideation or behaviour occurred during the placebo-controlled phase or apremilast-exposure period. During the placebo-controlled phase, two apremilast patients experienced an AE of depression; one had a history of depression and the other had dysthymia. Two additional AEs of depression were reported in the apremilast-exposure period; one patient had a history of depression. All four AEs of depression were not serious.
Throughout the study, markedly abnormal clinical laboratory values were infrequent and generally the result of single values outside the normal range (table 3).
No patients reported weight decrease as an AE during the study; 78.9% of apremilast patients remained within ±5% of their baseline weight. At the end of the 52-week period, mean weight loss for apremilast patients was −1.20 kg and 15.7% of apremilast patients had experienced >5% weight loss.
Discussion
ACTIVE was the first randomised controlled study to assess the onset of response to apremilast monotherapy in biological-naïve patients with active PsA. This study demonstrated that at week 2, many patients had clinical improvements across several PsA manifestations, including swollen and tender joints, enthesitis (among those with enthesitis at baseline), physical impairment and improvement in morning stiffness severity. Likewise, significant improvements in PsA measures at weeks 16 and 24 were observed with apremilast. Treatment response was maintained up to week 52 across measures for patients continuing apremilast and for placebo patients who switched to apremilast at week 16 or week 24.
These findings provide new data for apremilast, demonstrating that a proportion of patients experienced improvements in common symptoms of PsA at week 2. Additionally, the use of the GEI to assess peripheral entheseal sites in ACTIVE adds to our current knowledge of its effect on other entheseal sites, as measured by the Maastricht Ankylosing Spondylitis Enthesitis Score.
The PALACE studies evaluated apremilast in patients with several treatment failures (PALACE 1–3) and as a first-line treatment for DMARD-naïve patients (PALACE 4).4–7 Most (~70%) of the ACTIVE patient population had exposure to one csDMARD. Efficacy in this subpopulation was similar to that of the overall population in ACTIVE. These findings further support apremilast as a treatment option for patients with PsA across the spectrum of treatment experiences.
Apremilast was well tolerated in this biological-naïve PsA patient population; additionally, the overall safety profile in ACTIVE was found to be consistent with that observed in the PALACE studies.4–7 An important study objective was to further characterise the gastrointestinal AE of diarrhoea. Overall, fewer cases of protocol-defined diarrhoea (two or more watery stools/day) were observed versus non-defined reported events. This criterion is more inclusive than the WHO’s definition of diarrhoea of at least three loose or liquid stools/day. Diarrhoea AEs typically occurred within the first 4 weeks of treatment, were self-limiting, resolving within 15 days and usually did not require any major medical treatment.
Apremilast has a unique mechanism of action in modulating the expression of both pro-inflammatory and anti-inflammatory cytokines16; in ACTIVE, no evidence of increased incidence of serious or opportunistic infections and no cases of active tuberculosis with 52-week apremilast exposure were observed. Laboratory abnormalities were infrequent and showed no evidence of organ toxicity requiring specific monitoring. Safety results were consistent with the previous PALACE studies and provide additional characterisation of AEs of diarrhoea experienced during the placebo-controlled phase. The study design for ACTIVE allowed for immediate stopping of methotrexate without washout, which may be a desired option for some patients in routine clinical practice settings. Switching such as this happened seamlessly without any significant disease worsening/flares or tolerability issues.
Limitations
Several limitations should be considered when interpreting the study findings and comparing them with other apremilast clinical studies. The ACTIVE patient population had baseline heterogeneity regarding disease duration. Moreover, early escape was at the investigator’s discretion, which may be biased with apremilast availability on the market. Longer term findings may be biased because patients who did not respond to or tolerate treatment may be more likely to discontinue. ACTIVE did not evaluate dactylitis, skin and nail outcomes; however, apremilast’s impact on such outcomes has been assessed in the PALACE6 17 and Efficacy and Safety Trial Evaluating the Effects of Apremilast in Psoriasis (ESTEEM) studies.18 19 Additionally, this study did not include imaging to evaluate structural damage. Morning stiffness findings should be interpreted cautiously, as understanding of morning stiffness and PsA disease activity is limited.
Conclusions
For biological-naïve patients with active PsA, apremilast monotherapy resulted in early and sustained improvements across PsA manifestations, including swollen and tender joints, enthesitis and morning stiffness. No new safety concerns were observed. These results support the use of apremilast monotherapy in biological-naïve patients with PsA.
Acknowledgments
The authors would like to thank Thomas Rano, PhD, Suktae Choi, PhD, and Marla Hochfeld, MD, of Celgene Corporation for their contributions to the study. The authors received editorial support in the preparation of this report from Kristin Carlin, RPh, MBA, of Peloton Advantage, LLC, funded by Celgene Corporation. The authors, however, directed and are fully responsible for all content and editorial decisions.
References
Footnotes
Handling editor Tore K Kvien
Contributors All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. PN had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. Study conception and design: PN and ND. Acquisition of data: ND and DN. Analysis and interpretation of data: all authors. Statistical expertise: LT.
Funding This study was sponsored by Celgene Corporation.
Competing interests PN has received grant/research support and honoraria from Celgene Corporation. KO has received grant/research support from Celgene Corporation. JW has received non-financial support from Amgen Inc, Pfizer and UCB and has received personal fees from Celgene Corporation and Novartis. ND, DN and LT are employees of Celgene Corporation. JJG-R has received grant/research support from Roche and Schering-Plough and has served as a consultant to Bristol-Myers Squibb, Pfizer, Roche, Schering-Plough and UCB. JAA has received grant/research support from AbbVie Inc, Ardea Biosciences, Inc, AstraZeneca, Bristol-Myers Squibb, Celgene Corporation, Centocor, Galápagos, Genentech Inc, GlaxoSmithKline, Human Genome Sciences, Janssen, Eli Lilly and Company, Merck & Co, Mesoblast, Novartis Pharmaceuticals Corporation, Novo Nordisk, Pfizer, Roche, Sanofi-Aventis, Takeda Pharmaceuticals, UCB and Vertex Pharmaceuticals.
Patient consent Obtained.
Ethics approval Institutional review boards of the participating centres.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement All authors had full access to study data and had final responsibility for the decision to submit.
Collaborators The ACTIVE study investigators: Australia – Christopher Fong (Boxhill Hospital, Camberwell, VIC); Jane Zochling (Southern Clinical Research, Hobart, TAS); Peter Yousseff (Royal Prince Alfred Hospital, Camperdown, NSW); Paul Bird (St. George Private Hospital, Miranda, NSW); Nicholas Manolios (Westmead Hospital, Sydney, NSW); and Rob Will (Colin Bayliss Research/Teaching Unit, Victoria Park, WA). Canada – Ben Lasko and Melanie Mason (Manna Research, Toronto, ON); Jude Rodrigues (private practice, Windsor, ON); Alfred Cividino (private practice, Hamilton, ON); Majed Khraishi (Nexus Clinical Research, St. John’s, NL); Arthur Karasik (Arthur Karasik Medicine Professional Corp, Etobicoke, ON); and Louis Bessette (Groupe de Recherche en Maladies Osseuses Inc., Quebec, QC). Czech Republic – Petr Vitek (PV Medical Sro, Zlin); Zuzana Stejfova and Alena Ticha (Revmatologicka Ambulance, Praha); and Katerina Jarosova (Revmatologicky Ustav, Praha). Estonia – Airi Poder (Clinical Research Centre Ltd, Tartu); Andres Pille (East Tallinn Central Hospital, Tallinn); and Jaak Tälli (Innomedica, Tallinn). Hungary – László Sámson (MÁV Kórház és Rendelőintézet, Budapest); Istvan Szombati (Qualiclinic Kft, Budapest); Kiss Csaba (Csolnoky Ferenc Kórház, Veszprém); and Szántó Sándor (Debreceni Egyetem Orvos, Debreceni). New Zealand – Daniel Ching (Timaru Hospital, Timaru); Rajiv Gupta (Middlemore Hospital, Auckland); and Doug White (Rheumatology Clinic, Auckland). Romania – Mariana Pavel (Sf. Apostol Andrei Emergency Clinical County Hospital, Galați); Violeta Bojinca (Sf. Maria Clinical Hospital, Bucharest); Silvia Bojin (Covamed Serv SRL, Sfântu Gheorghe); and Simona Rednic (Cluj-Napoca Emergency Clinical County Hospital, Cluj-Napoca). Russia – Valentin Oleynikov (Penza Regional Clinical Hospital, Penza); Leysan Myasoutova (City Clinical Hospital #7, Kazan); Nikolay Korshunov (Yaroslavl Reg. Clinical Hospital, Yaroslavl); and Diana Krechikova (Dept. Hospital at Smolensk, Smolensk). Spain – Francisco Blanco (Complejo Hospitalario Universitario, Coruña); Antonio Fernandez-Nebro (Hospital General de Málaga, Málaga); Federico Díaz-Gonzalez (Hospital Universitario de Canarias, Santa Cruz de Tenerife); Eduardo Angulo (Hospital de Basurto – Osakidetza, Basurto); Emilio Mola (Hospital Universitario La Paz, Madrid); Jordi Gratacos Masmitja (Corporació Sanitària Parc Taulí, Barcelona); Jesús Rodriguez (Hospital Universitari de Bellvitge, l’Hospitalet de Llobregat); and Agusti Sellas Fernandez (Hospital Universitari Vall d’Hebron, Barcelona). United States – Monika Mohan (Advanced Rheumatology, Lansing, MI); Richard Olson (Rockford Orthopedic Associates, Ltd, Rockford, IL); Carmen Perez-Masuelli (The Clinical Research Institute of Houston, Houston, TX); Jeffrey Alloway (Physicians East, PA, Greenville, NC); Jeffrey Alper (private practice, Naples, FL); Alan J. Kivitz (Altoona Center For Clinical Research, Altoona, PA); Miriam Lara (Palmetto Medical Research, Hialeah, FL); Vipul Joshi (Bay Area Arthritis and Osteoporosis, Brandon, FL); Roger Diegel (Glacier View Research Institute, Kalispell, MT); Tina Bunch (Austin Regional Clinic, Austin, TX); Alfred Felber (Heartland Clinical Research Inc., Augusta, KS); Michael Calmes (Arthritis and Osteoporosis Associates, LLP, Lubbock, TX); David McLain (Achieve Clinical Research, LLC, Birmingham, AL); Robert Holmes (Piedmont Medical Research, LLC, Winston-Salem, NC); John Carter (University of South Florida, Tampa, FL); Farrukh Zaidi (Suncoast Clinical Research, New Port Richey, FL); Maria Greenwald (Desert Medical Advances, Palm Desert, CA); Michael Burnette (Health Point Medical Group, Brandon, FL); Ramesh Gupta (private practice, Memphis, TN); Charles Bradley Franz (Mountain State Clinical Research, Bridgeport, WV); John Cush (Baylor Research Institute, Dallas, TX); Craig Wiesenhutter (Coeur d’Alene Arthritis Clinic, Coeur d’Alene, ID).
Author note The results from this study have been published in abstract form: Nash P et al. Early onset of efficacy with apremilast monotherapy in biologic-naïve patients with active psoriatic arthritis: a phase IIIB, randomized, controlled trial [abstract 1703]. Arthritis Rheumatol 2016;68(Suppl 10):2133–4, and Nash P et al. Early onset of efficacy with apremilast monotherapy in biologic-naïve patients with active psoriatic arthritis: a phase IIIB, randomized, controlled trial [abstract OP0219]. Ann Rheum Dis 2017;76(Suppl 2):143.