Article Text

Abstract
Objectives Efficacy, safety and immunogenicity results from the phase III study of SB2, a biosimilar of reference infliximab (INF), were previously reported through 54 weeks. This transition period compared results in patients with rheumatoid arthritis (RA) who switched from INF to SB2 with those in patients who maintained treatment with INF or SB2.
Methods Patients with moderate to severe RA despite methotrexate treatment were randomised (1:1) to receive SB2 or INF at weeks 0, 2 and 6 and every 8 weeks thereafter until week 46. At week 54, patients previously receiving INF were rerandomised (1:1) to switch to SB2 (INF/SB2 (n=94)) or to continue on INF (INF/INF (n=101)) up to week 70. Patients previously receiving SB2 continued on SB2 (SB2/SB2 (n=201)) up to week 70. Efficacy, safety and immunogenicity were assessed up to week 78.
Results Efficacy was sustained and comparable across treatment groups. American College of Rheumatology (ACR) 20 responses between weeks 54 and 78 ranged from 63.5% to 72.3% with INF/SB2, 66.3%%–69.4% with INF/INF and 65.6%–68.3% with SB2/SB2. Treatment-emergent adverse events during this time occurred in 36.2%, 35.6% and 40.3%, respectively, and infusion-related reactions in 3.2%, 2.0% and 3.5%. Among patients who were negative for antidrug antibodies (ADA) up to week 54, newly developed ADAs were reported in 14.6%, 14.9% and 14.1% of the INF/SB2, INF/INF and SB2/SB2 groups, respectively.
Conclusions The efficacy, safety and immunogenicity profiles remained comparable among the INF/SB2, INF/INF and SB2/SB2 groups up to week 78, with no treatment-emergent issues or clinically relevant immunogenicity after switching from INF to SB2.
Trial registration number NCT01936181; EudraCT number: 2012-005733-37.
- anti-tnf
- dmards (biologic)
- rheumatoid arthritis
- tnf-alpha
- treatment
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
The introduction of biosimilars has significantly impacted medical practice and the pharmaceutical industry.1 2 While biologicals are effective, they are also expensive, thus creating inequity by limiting their accessibility to patients and countries that can afford them.3 4 Biosimilars have the potential to improve access to treatment by reducing the financial burden on healthcare systems.5
While from a physician’s perspective, biosimilars may be considered akin to chemical generics, making identical copies of biologicals is not technically feasible, and biosimilars undergo a more comprehensive regulatory pathway. This includes preclinical quality analysis, pharmacokinetic and pharmacodynamic assessments and phase III clinical evaluation, which is usually conducted in a randomised, double-blind fashion in at least one of the originator’s indications.6 7 Clinical trials of biosimilars are usually parallel-arm equivalence studies, with the primary aim to test that the biosimilar has equivalent efficacy and comparable safety to the reference product.6–10
An important issue surrounding biosimilars that cannot be tested by this approach is whether patients can be switched from the originator without major concerns.1 Because the main objective of biosimilars is to reduce drug costs and make biologicals more affordable to a larger population,11 switching patients from the original biological to a biosimilar is a likely consideration in clinical practice to capitalise on the cost reduction. However, as previously mentioned, biosimilars are not identical to their original counterparts. Additionally, biologicals commonly have issues with immunogenicity, which can be associated with decreased efficacy and, in some cases, with adverse events (AEs).12 Therefore, data regarding switching from originators to biosimilars are desirable to strengthen the demonstration of biosimilarity.
SB2 (Samsung Bioepis, Incheon, Republic of Korea) and reference infliximab (INF; Remicade, Janssen Biotech, Horsham, Pennsylvania, USA) have been shown to have equivalent efficacy and comparable structure, function, pharmacokinetic parameters, immunogenicity and safety.8 13 14 SB2 was approved in the USA on 21 April 2017 and has also been approved in Norway, Liechtenstein, Iceland and Australia, in addition to having been approved in the European Union15 and Korea.16 The clinical efficacy and safety results of SB2 for the treatment of rheumatoid arthritis (RA) were previously reported, up to 54 weeks, based on a phase III equivalence study conducted using the aforementioned parallel-arm design.8 17 The objectives of the present transition-extension period (described as transition period hereafter) of the phase III study were to investigate whether individuals on INF could be readily switched to SB2 without major concerns and whether comparable efficacy, safety and immunogenicity were maintained after the switch when compared with both ongoing reference INF as well as SB2.
Methods
Methods for the initial randomised, double-blind period of this multinational, multicentre, parallel group study (weeks 0–54) have been previously described.8 17 The study originally enrolled patients 18–75 years of age diagnosed with moderate to severe RA (1987 American College of Rheumatology (ACR) criteria) despite methotrexate therapy. The methods below focus on the transition period (weeks 54–78).
Patients
Those who completed the week 54 visit of the randomised, double-blind period and were willing to participate were eligible for the transition period. Patients who experienced any significant medical condition(s) during the randomised, double-blind period, such as the occurrence of a serious AE (SAE) or intolerance of SB2 or INF, and who were determined to be unfit for further treatment were excluded.
Study design
Patients were initially randomised (1:1) to receive either SB2 or INF at weeks 0, 2 and 6 and then every 8 weeks thereafter until week 46 (randomised, double-blind period). The protocol was amended during this period to accommodate the transition design. At week 54, enrolled patients in the INF group were rerandomised (1:1) to either transition (switch) to SB2 (INF/SB2) or to continue on INF (INF/INF) up to week 70 (transition period, figure S1 in online supplementary appendix). Patients in the SB2 group continued to receive SB2 up to week 70 (SB2/SB2) but followed the randomisation procedure to maintain double-blind status. The final visit was at week 78. An interactive web response system was used for randomisation and treatment allocation.8
Supplementary file 1
Treatment with SB2 or INF was initiated at an intravenous dose of 3 mg/kg at week 0. The dose could have been increased stepwise by 1.5 mg/kg, up to a maximum of 7.5 mg/kg, starting at week 30 and every 8 weeks thereafter if the patient’s RA symptoms were not well controlled by the existing dose. At the time of switching to SB2 from INF (or continuing INF or SB2 in the other arms), the dosing schedule continued from the last dose applied before switching (ie, week 54). An oral or parenteral stable dose of methotrexate (10–25 mg/week) was taken with folic acid (5–10 mg/week) throughout the study. No other disease-modifying antirheumatic drugs were permitted. Paracetamol, antihistamines and/or corticosteroids were allowed as premedications at the investigator’s discretion to prevent infusion-related reactions.
Assessments
At each clinic visit, efficacy was evaluated by ACR response rates (ACR20, ACR50 and ACR70), disease activity score based on a 28-joint count (DAS28 score) and European League Against Rheumatism (EULAR) responses. Clinical Disease Activity Index (CDAI) and Simplified Disease Activity Index (SDAI) scores18 were calculated post hoc. Safety was monitored throughout the study by evaluation of treatment-emergent AEs (TEAEs), SAEs and AEs of special interest (serious infections or tuberculosis); latent tuberculosis was monitored with QuantiFERON Gold blood tests at weeks 54 and 78. Immunogenicity was assessed by the development of serum antidrug antibodies (ADAs) and neutralising antibodies (NAb) among who were ADA positive.8
Statistical analysis
Sample size and power calculations based on the primary endpoint of the study (ACR20 response at week 30) were previously described.8 All results obtained during the transition period were analysed using descriptive statistics. Efficacy results were based on the extended full analysis set (Ex-FAS), which follows the intent-to-treat principle and comprises available data (ie, no imputation) in all patients who were rerandomised at week 54 and who received at least one dose of SB2 or INF during the transition period. To evaluate efficacy changes over the entire duration of the study in the three treatment groups, a retrospective analysis of efficacy was performed in the Ex-FAS population from week 54 back to week 0. AEs and immunogenicity were analysed in the extended safety set (Ex-SAF), which comprised all patients who received at least one dose of SB2 or INF during the transition period. Analyses were performed using SAS V.9.2.
Results
Patients
The study started in August 2013, and the transition period was completed in August 2015. Patient disposition is shown in figure 1. At week 54, 396 patients were rerandomised to receive SB2/SB2 (n=201), INF/SB2 (n=94) or INF/INF (n=101) and were included in this analysis. The majority of patients in each treatment group completed the transition period (92.5%, 93.6% and 95.0%, respectively). The number and pattern of withdrawals were comparable among the three treatment groups.

Patient disposition of the study population. *Percentages of patients completed and discontinued are based on the number of patients rerandomised at week 54. Note: eight patients’ data from sites in eastern Ukraine were excluded from the analysis because of regional issues (n=4 in SB2, n=4 in INF). INF, reference infliximab.
Patient demographics and disease characteristics of the rerandomised population were well balanced among the three treatment groups at baseline, and disease characteristics were also comparable at the time of rerandomisation (table 1). At weeks 54, 62 and 70, the proportion of patients treated with 3, 4.5, 6 or 7.5 mg/kg of investigational product was similar in the INF/SB2, INF/INF and SB2/SB2 groups (table S1 in online supplementary appendix).
Patient demographics and disease characteristics of the rerandomised population at baseline (A and B) and rerandomisation (C)
Efficacy
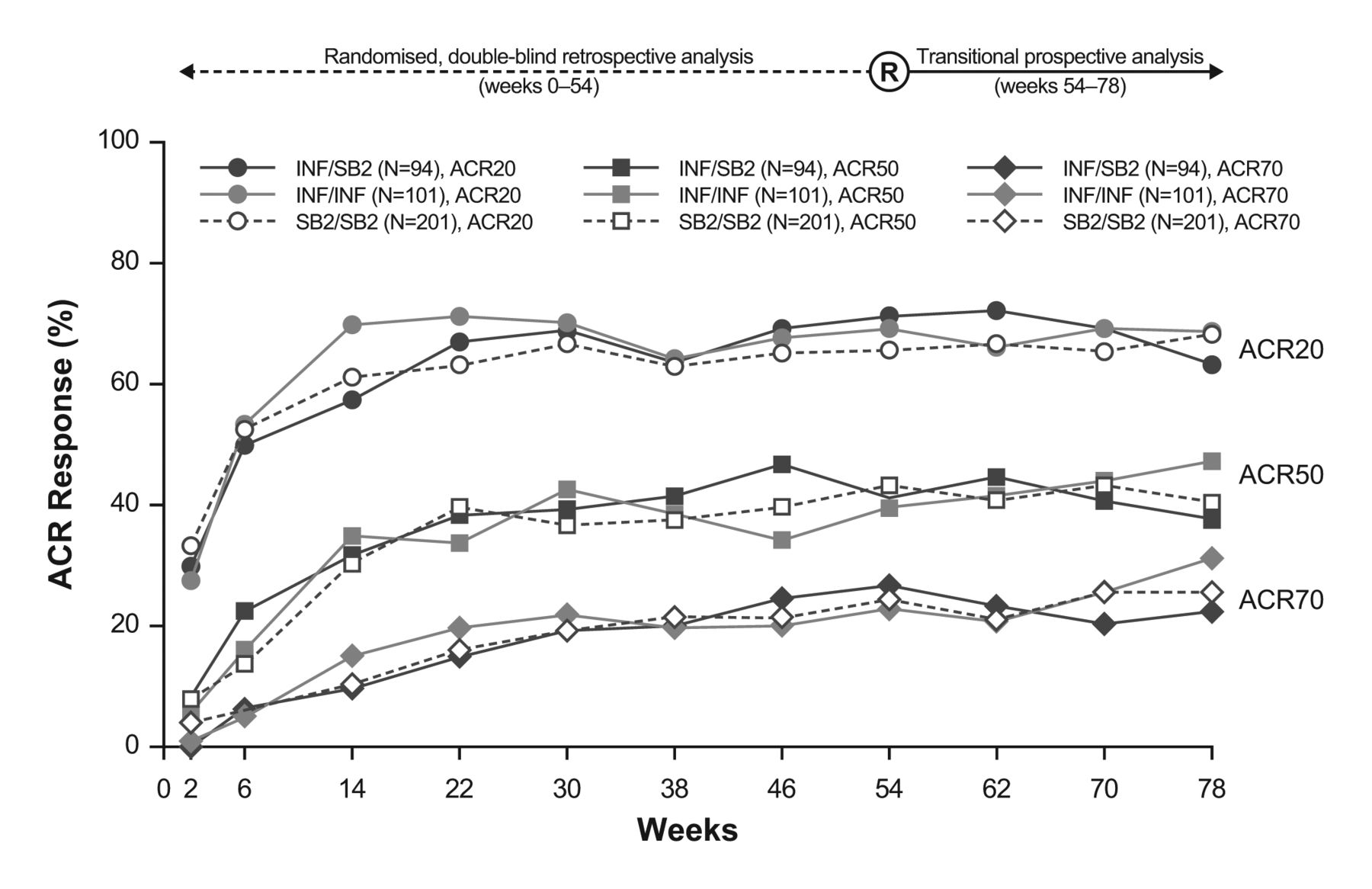
The time–response pattern of mean DAS28, SDAI and CDAI in this transition study population is shown in figure 2. The pattern of disease activity improvement was highly similar among the three treatment groups during the entire study period. Figure 3 and table S2 (see online supplementary appendix) show the ACR20, 50 and 70 response rates, which were comparable across the double-blind randomised and transition period. While a somewhat higher variance was observed, especially in patients initially treated with INF who either transitioned to SB2 or continued INF, this pattern was already evident during the pretransition period before rerandomisation (assessed retrospectively), and the overall pattern did not deviate meaningfully during the period after the switch. The proportion of EULAR responses classified as good or moderate was comparable at week 78 across the treatment groups (good: 32.9%–35.6% of patients; moderate: 50.5%–51.8% of patients; figure S2 in online supplementary appendix).

Mean disease activity score based on a 28-joint count (DAS28 (ESR)) (A), Clinical Disease Activity Index (CDAI) score (B) and Simplified Disease Activity Index (SDAI) score (C) up to week 78. ESR, erythrocyte sedimentation rate; INF, reference infliximab.

American College of Rheumatology (ACR) responses up to week 78. The responses before week 54 are retrospective analyses based on the extended full analysis set. For the actual percentages, please refer to online supplementary appendix table S2. INF, reference infliximab.
When considering efficacy after dose increase of INF was permitted (ie, week 30 and thereafter), the efficacy response pattern was comparable among the three treatment groups, both in patients who had received at least one dose increment and in those who did not receive any dose increments (figure S3 in online supplementary appendix). Patients who needed at least one dose increment of INF or SB2 had experienced lower response rates than patients who did not have a dose increment, and patients who had a dose increment experienced an increase in efficacy across treatment groups. Such response patterns were generally consistent before and after rerandomisation (week 54). At week 78, patients who did not receive any dose increment and who had transitioned from INF to SB2 had a numerically lower ACR20 response rate than those who did continue treatment with either INF or SB2 throughout the entire study (figure S3 in online supplementary appendix). Thus, some variance in response pattern was observed in the INF/SB2 treatment group, which is thought to be a reflection of the overall efficacy pattern seen in figure 3.
Safety
The overall incidence of TEAEs reported during the transition period in the Ex-SAF population was comparable in each treatment group (table 2). The most commonly reported TEAEs during this period were latent tuberculosis, nasopharyngitis and RA (worsening); there were no deaths or new cases of active tuberculosis during the transition period. Three cases of malignancy were reported during the transition period: lip and/or oral cavity cancer and basal cell carcinoma in the INF/SB2 group and papillary thyroid cancer in the INF/INF group. Rates of serious TEAEs, serious infections and infusion-related reactions were low and comparable across the three treatment groups (table 2). There were four serious infections reported: two events in the INF/SB2 treatment group of arthritis bacterial and haematoma infection, one event in the INF/INF treatment group of respiratory tract infection and one event in the SB2/SB2 treatment group of urosepsis.
Summary of safety profile during the transition period
Immunogenicity
The incidence of overall ADA after transition and newly developed ADA after transition was comparable in the three treatment groups (figure 4). The incidence of overall positive ADA during the transition period among patients with overall negative ADA up to week 54 was 14.6% for INF/SB2, 14.9% for INF/INF and 14.1% for SB2/SB2 (NAb 33.3%, 71.4% and 63.6%) indicating that immunogenicity after switching from INF to SB2 was similar to that from continuing either INF or SB2.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Incidence of immunogenicity during the transition period. *Patients having at least one positive ADA result during the transition-extension period among all patients regardless of prior ADA result up to week 54 (n=94 in INF/SB2, n=101 in INF/INF, n=201 in SB2/SB2). †Patients having at least one positive ADA result during the transition period among patients with overall negative ADA results up to week 54 (n=41 in INF/SB2, n=47 in INF/INF, n=78 in SB2/SB2). NAb was measured among ADA positive subjects (n=6, 7 and 11). ADA, antidrug antibody; NAb, neutralising antibody; INF, reference infliximab.
Discussion
Here we report results from the transition period of the phase III study of the INF biosimilar, SB2, in patients with moderate-to-severe RA despite methotrexate treatment. Our main goal was to demonstrate clinical comparability of switching from INF to SB2 with both ongoing reference INF as well as SB2. This type of comparative approach may be considered unique in INF biosimilar studies done hitherto; for example, switching of INF to CT-P13 was compared with continuing CT-P13, but not with a parallel, continuing reference INF arm.19
When switched from INF to SB2, there was no clinically meaningful difference in terms of efficacy, safety and immunogenicity compared with the INF/INF group. Likewise, the SB2/SB2 group also maintained long-term efficacy, safety and immunogenicity, again comparable with that of the long-term INF/INF group or the INF/SB2 group. Even dose increment patterns after week 54 were comparable among the three treatment groups, with a similar efficacy response. These results are consistent with our previous reports of SB28 17 but provide additional insight on switching and longer term treatment. Also, our study is unique among INF biosimilar studies in that it employed a switching design and continued with dose increments, both of which can be situations encountered in the clinical setting.
Our data showing that switching from originator to biosimilar is safe and effective are corroborated by the recent observations in the NOR-SWITCH and DANBIO study.19 20 In the NOR-SWITCH study, efficacy and safety in patients with multiple diagnoses who were switched from reference INF to biosimilar CT-P13 were compared with those maintaining reference INF, revealing similar results, while in the DANBIO study, prior INF-receiving patients were non-medically switched to CT-P13 due to national policy yet maintaining similar disease activity compared with historical INF data. In both studies, comparison with a continuing biosimilar could not be tested, because it had not been available prior to initiation of the trial or non-medical switch.19 20
Recently, various study designs have been proposed to address the issue of biosimilar switching. Early switching designs employed a total group switch in which the originator treatment group was switched entirely to the biosimilar and compared with the ongoing biosimilar treatment group.21 22 Others employed a multiple switch design, switching back and forth in both the originator and biosimilar treatment groups.23 Our study design split the originator treatment arm into two groups and switched one of these groups to the biosimilar. While it is not clear which design is best for assessing biosimilar switching, our study allows simultaneous comparison of the switched group with both the ongoing originator and biosimilar groups, respectively, as mentioned previously.
Another important factor in switching designs is maintenance of study blinding. Because patients might exhibit different attitudes when becoming aware of receiving a biosimilar, this could potentially affect the study outcomes. Such ‘nocebo’ effects have been reported with chemical generic drug switching.24 To avoid these effects, our study was fully blinded throughout, even including mock-randomisation procedures for the SB2/SB2 treatment group that did not change during the entire study period, thus minimising possible bias.
As a limitation of our study, because the INF population was split into two groups, the sample size for each treatment group decreased by half. This may have increased the potential for greater variation in clinical outcomes, possibly making comparisons between the treatment groups somewhat more difficult. This is suggested by the wider efficacy fluctuations seen in the INF/SB2 and INF/INF groups compared with the more stable pattern seen in the SB2/SB2 group; however, this variability already existed in the pretransition period on post hoc analysis. Thus, it is reassuring that despite such potential variations, the efficacy, safety and immunogenicity outcomes were comparable among the three treatment groups.
Conclusions
SB2, an INF biosimilar, maintained comparable efficacy, safety and immunogenicity up to 78 weeks, even after switching from the originator INF. Our results suggest that the clinical profile of SB2, when administered long term or when switched from INF, is comparable with INF.
Acknowledgments
The authors would like to thank the patients who were involved in this study, the study personnel who made this work possible and the study investigators: Bosnia and Herzegovina: Sokolovic S; Bulgaria: Dimitrov E, Geneva-Popova M, Mihaylova M, Toncheva A, Penev D and Oparanov B; Czech Republic: Galatikova D, Vitek P and Janska L; Korea, Republic of: Shim SC, Kang YM, Kim HA, Kim SK, Lee S-H, Bae S-C, Kim J, Kwok S-K, Lee YJ and Lee S-K; Latvia: Kadisa A, Mihailova A, Saulite-Kandevica D and Saleniece S; Lithuania: Milasiene R, Arstikyte I and Basijokiene V; Poland: Kolczewska A, Stasiuk B, Grabowicz-Wasko B, Leszczynski P, Ruzga Z, Rychlewska-Hanczewska A and Hilt J; Romania: Mirea G, Pavel M, Ieremia G and Tanasescu C; Ukraine: Rekalov D, Zhdan V, Povoroznyuk V, Ter-Vartanian S, Stanislavchuk M, Golovchenko O, Tseluyko V, Yagensky A, Iaremenko O and Shevchuk S; and the study team: Ilsun Hong (Samsung Bioepis). Editorial support for development of this manuscript was provided by Michael S. McNamara at C4 MedSolutions, LLC (Yardley, Pennsylvania, USA), a CHC Group company, and funded by Samsung Bioepis Co., Ltd.
References
Footnotes
A portion of this manuscript, including results up to week 78, has been previously presented at EULAR 2016 (Poster # FRI0162) and published in abstract form as follows: Smolen JS, Choe J-Y, Prodanovic N, et al. Comparable safety and immunogenicity and sustained efficacy after transition to SB2 (an infliximab biosimilar) vs ongoing infliximab reference product in patients with rheumatoid arthritis: results of phase III transition study. Ann Rheum Dis 2016;75(Suppl 2):488.
Handling editor Tore K Kvien
Contributors YL and YHR contributed to study conception and design, acquisition of data, analysis and interpretation of data, drafting the manuscript and revising it critically for important intellectual content and final approval of the version to be published. JSS contributed to acquisition of data, analysis and interpretation of data, drafting the manuscript and revising it critically for important intellectual content and final approval of the version to be published. J-YC, NP, JN, IS, ED, AB, RY, MM, WP, HC, KJ-R and AZ contributed to acquisition of data, drafting of the manuscript and revising it critically for important intellectual content and final approval of the version to be published. All coauthors contributed significantly to the writing of the manuscript.
Funding This study was funded by Samsung Bioepis Co., Ltd.
Competing interests JSS reports receiving grant/research support from AbbVie, Janssen, MSD, Pfizer, Roche and UCB; consultant for AbbVie, Amgen, AstraZeneca, Astro-Pharma, Celgene, GSK, Janssen, Lilly, Medimmune, MSD, Novartis-Sandoz, Novo Nordisk, Pfizer, Roche, Samsung Bioepis, Sanofi and UCB. J-YC reports receiving grant/research support and consultant fees from Samsung Bioepis. AB reports receiving grant/research support from AbbVie and Samsung Bioepis. NP, JN, IS, ED, RY, MM, WP, HC, KJ-R and AZ report receiving grant/research support from Samsung Bioepis. YL and YHR are employees of Samsung Bioepis and own stocks in Samsung Biologics.
Ethics approval Ethics approval was received from each national regulatory agency and central or local ethical committee.
Provenance and peer review Not commissioned; externally peer reviewed.