Article Text

Abstract
The past three decades have witnessed remarkable advances in our ability to target specific elements of the immune and inflammatory response, fuelled by advances in both biotechnology and disease knowledge. As well as providing superior treatments for immune-mediated inflammatory diseases (IMIDs), such therapies also offer unrivalled opportunities to study the underlying immunopathological basis of these conditions.
In this review, we explore recent approaches to the treatment of IMIDs and the insights to pathobiology that they provide. We review novel biologic agents targeting the T-helper 17 axis, including therapies directed towards interleukin (IL)-17 (secukinumab, ixekizumab, bimekizumab), IL-17R (brodalumab), IL-12/23p40 (ustekinumab, briakinumab) and IL-23p19 (guselkumab, tildrakizumab, brazikumab, risankizumab, mirikizumab). We also present an overview of biologics active against type I and II interferons, including sifalumumab, rontalizumab, anifrolumab and fontolizumab. Emerging strategies to interfere with cellular adhesion processes involved in lymphocyte recruitment are discussed, including both integrin blockade (natalizumab, vedolizumab, etrolizumab) and sphingosine-1-phosphate receptor inhibition (fingolimod, ozanimod). We summarise the development and recent application of Janus kinase (JAK) inhibitors in the treatment of IMIDs, including first-generation pan-JAK inhibitors (tofacitinib, baricitinib, ruxolitinib, peficitinib) and second-generation selective JAK inhibitors (decernotinib, filgotinib, upadacitinib). New biologics targeting B-cells (including ocrelizumab, veltuzumab, tabalumab and atacicept) and the development of novel strategies for regulatory T-cell modulation (including low-dose IL-2 therapy and Tregitopes) are also discussed. Finally, we explore recent biotechnological advances such as the development of bispecific antibodies (ABT-122, COVA322), and their application to the treatment of IMIDs.
- autoimmune diseases
- autoimmunity
- DMARDs
- treatment
Statistics from Altmetric.com
Introduction
Rapid progress in both disease knowledge and biotechnology over the past three decades has led to an increasingly diverse armamentarium of therapies for immune-mediated inflammatory diseases (IMIDs). As well as providing better and more focused therapies, these novel approaches can provide unique insights into disease pathogenesis or, indeed the complications of therapy. An early example was the recognition of the central importance of tumour necrosis factor-α (TNF-α) in granuloma maintenance, and hence protection against reactivation of latent tuberculosis.1 In this review, we discuss recent approaches to the treatment of IMIDs, with a particular focus on biological and biotechnological advances, and examine the insights that they provide.
The Th17 axis
CD4+ T cells sit at the interface between innate and adaptive immunity, and are considered the orchestrators of the adaptive immune response. Early studies of CD4+ T-cell biology described two mutually exclusive phenotypes, T-helper (Th)1 and Th2. Th1 cells promote cellular immunity against intracellular pathogens via the release of cytokines such as interferon gamma (IFN-γ), whereas Th2 cells promote humoral immunity and the response to helminth infections via the production of interleukin (IL)-4, IL-5 and IL-13.2 Th1 cells were initially regarded as the drivers of many IMIDs, including rheumatoid arthritis (RA), although both animal and human data suggested that they were not always essential, catalysing the search for alternative subsets.3 Th17 cells appeared to fill this gap, at least in some diseases. IL-17, one of the cytokines produced by this subset, is a potent pro-inflammatory cytokine which, together with TNF-α and IL-1β, recruits neutrophils as well as inhibiting chondrocyte metabolism and promoting osteoclastogenesis.2 Since their discovery, Th17 cells have been implicated in a variety of IMIDs including RA, psoriasis, psoriatic arthritis (PsA), ankylosing spondylitis (AS) and inflammatory bowel disease.4 Blocking the Th17 axis, either by inhibiting IL-17 directly or via preventing Th17 cell differentiation, is now an area of intense therapeutic development (figure 1).5

Strategies to block IL-17
IL-17 comprises a family of six homologous cytokines (IL-17A to F), of which IL-17A is the most abundant, most potent and best characterised.6 Homodimers and heterodimers of IL-17 cytokines signal via dimeric IL-17 receptors, of which there are five identified subunits (IL-17RA to E).6 The precise binding affinity, cellular distribution and downstream action of the various IL-17 receptors are yet to be fully elucidated. Nevertheless, it is apparent that blockade of different IL-17 cytokines or their receptors can yield quantitatively and qualitatively different therapeutic responses.
Secukinumab is a fully human monoclonal antibody (mAb) against IL-17A, and is effective in the treatment of plaque psoriasis, PsA and AS,7 although has proved disappointing in the treatment of RA.8 Ixekizumab, a humanised anti-IL-17A mAb, has similarly shown efficacy to date in psoriasis9 and PsA.10 In PsA trials, articular efficacy of IL-17 blockade is similar to TNF blockade (and superior to effects in RA), whereas skin efficacy is clearly superior to that of TNF blockade.9 More recently bimekizumab, a humanised mAb directed against a homologous epitope shared between both IL-17A and IL-17F, has shown promising results in early phase clinical trials in psoriasis11 and PsA.12 Other biologics that neutralise both IL-17A and F, including true bispecific antibodies (see the ‘Newer technologies’ section), are currently in development (table 1). Blockade of IL-17 is associated with the development of candidiasis, which is not generally severe, but highlights the importance of IL-17 in fungal defence.
Development of biologics active against Th17 pathway targets
In addition to cytokine neutralisation, IL-17 inhibition can be achieved by blocking the IL-17 receptor. Brodalumab is a human mAb against IL-17RA, which is required for formation of the dimeric receptors necessary for IL-17A, IL-17F, IL-17A/F, IL-17C and IL-17E (IL-25) signalling.13 Brodalumab thus exerts a much broader blockade of IL-17 signalling than the targeting of specific cytokines (figure 1). Nevertheless, the inhibition of IL-17E, which promotes a Th2 response while potentially inhibiting Th17 differentiation in mice,14 and in human IBD15 and RA,16 could be therapeutically counterproductive in some disease settings. As discussed by Patel and Kuchroo,3 this may explain the lack of effect of brodalumab in RA,17 despite modest efficacies of secukinumab18 and ixekizumab.19 Brodalumab is licensed for the treatment of psoriasis in Japan and the USA, and will shortly receive European marketing authorisation.
Similar to RA, IL-17 blockade appears to have limited efficacy in non-infective uveitis.3 These observations highlight that the presence of a cytokine in diseased tissue does not necessarily equate to an irreplaceable role in pathogenesis. Furthermore, both secukinumab20 and brodalumab21 have been demonstrated to worsen Crohn’s disease (CD). Thus, in contrast to its pro-inflammatory role in other diseases and locations, IL-17A may function as a negative regulator of immunity in the gut mucosa, perhaps via interaction with fungal elements of the intestinal microbiome.22 23 This observation is important in view of the clinical overlap between seronegative IMIDs, for example, trials of IL-17 blockade in psoriasis were associated with haemorrhagic diarrhoea as an adverse reaction.24 In addition, concerns surrounding a possible association between brodalumab and suicidal ideation have hampered the development of this drug,25 although these concerns may have been overstated.26
Th17 differentiation and the IL-12 superfamily
Central to the polarisation of naïve CD4+ T cells to distinct effector phenotypes are members of the IL-12 cytokine superfamily, namely IL-12, IL-23, IL-27 and IL-35.27 These cytokines and their receptors also exist as dimers with considerable sequence homology between subunits, although with often opposing roles in immunity. Increasing knowledge of the constituent components of this family, particularly IL-12 and −23, has brought opportunities for therapeutic intervention aimed at blocking pathogenic Th17 differentiation28 (figure 1, table 1).
Ustekinumab is a fully human mAb against the p40 subunit common to IL-12 and IL-23, licensed for the treatment of psoriasis, PsA and CD.29 It has also shown benefit in AS in an open-label study30 and a separate post hoc pooled analysis.31 Intriguingly, and in direct contrast to IL-17A blockade, ustekinumab is effective in CD with evidence for a prolonged benefit following a single infusion.32 33 Paradoxically, ustekinumab was inferior to secukinumab in moderate-to-severe psoriasis with a comparable safety profile.34 These contrasting observations demonstrate first that blocking Th17 differentiation via IL-23 inhibition is mechanistically distinct from the blockade of IL-17A itself, and second that the relative effects differ between diseases.
It is also possible to selectively inhibit IL-23 by targeting its unique p19 subunit, and numerous such mAbs are in development for the treatment of psoriasis and IBD (figure 1, table 1). Purported advantages of selective IL-23 inhibition over dual IL-12/IL-23 blockade include the potential for less severe infections and lower malignancy risk; indeed, postmarketing data in psoriasis suggest an increased risk of non-melanoma skin cancer with ustekinumab, which may be a consequence of inhibition of IL-12-mediated cellular immunity.35 Whether these theoretical advantages translate to clear benefits in the long term awaits confirmation.
Head-to-head trials in psoriasis
As illustrated above, head-to-head trials in psoriasis have been particularly illuminating with regard to the relative dominance of pathogenic pathways in this condition. For example, both IL-17A blockade with ixekizumab9 and IL-23 blockade with guselkumab36 are superior to TNF-α blockade (with etanercept and adalimumab, respectively). In other trials, IL-17A blockade with secukinumab,34 IL-17R blockade with brodalumab37 and IL-23p19 blockade with risankizumab38 all appear superior to IL-12/23p40 blockade with ustekinumab. IL-17A, IL-17A/F, IL-17R and IL-23p19 blockade look similarly effective in these various trials, although, as in CD, IL-23p19 blockade appears to have particularly long-lasting efficacy.
The reason for the distinct effects of IL-12/23p40 versus IL-17 blockade is not immediately apparent, particularly the contrasting effects in different diseases. However, Th17 cells produce cytokines other than IL-17A (eg, IL-17F and IL-22), and IL-17A is also produced by cellular subtypes other than Th17 cells. These are less influenced by IL-23 family signalling, and in some environments (eg, the gut), IL-17 may even have regulatory functions.4 23 Furthermore, IL-12/23p40 blockade also inhibits IL-12 signalling, a pro-Th1 cytokine which plays a key role in the pathogenesis of CD.39 It is therefore apparent that simultaneous blockade of multiple cytokines, or blockade of the same immune axis at different hierarchical levels, can produce different outcomes dependent on the tissue and disease context in which it is deployed.
Summary: blocking the IL-17 axis—what have we learnt?
In summary, IL-17 is a key pathogenic cytokine in multiple IMIDs. However, its mere presence does not necessarily imply a dominant pathogenic role. Furthermore in a single condition, such as PsA, its relative role may vary between tissues. Lastly, blocking the axis at distinct levels can have differing effects, with distinct hierarchies in different diseases.
The type I IFN axis
IFNs are a family of potent immunostimulatory cytokines that are broadly divided into three subtypes: type I (IFN-α, β, ε, κ and ω), type II (IFN-γ) and the newly characterised type III (IFN-λ)40 (figure 2). Of all the type I IFNs (IFN-I), IFN-α is the most abundant and best characterised, and exists in 13 distinct although homologous subtypes.40 IFN-I production is tightly regulated such that levels are virtually undetectable in health. However, during pro-inflammatory states, such as viral infections, IFN-I is rapidly produced in large quantities.41 Especially notable in their propensity to secrete IFN-I are plasmacytoid dendritic cells (pDCs), which abundantly express intracellular pattern recognition receptors such as toll-like receptor (TLR)-7 and TLR-9.42 On ligation of the IFN-I receptor (IFNAR), IFN-I induces the upregulated expression of a stereotypical set of genes, known as IFN-stimulated genes (ISGs).40 The effects of IFN-I are vigorously pro-inflammatory and include dendritic cell maturation and activation, Th1 and Th17 polarisation, reduced regulatory T cells (Treg) function and increased B-cell activation and subsequent antibody production.41

Overview of biologics targeted against interferon (IFN) pathways. GAS, interferon-γ activated site; IRF9, interferon regulatory factor 9; ISREs, interferon-stimulated response elements; JAK1/2, Janus kinase 1/2; pDC, plasmacytoid dendritic cell; STAT1/2, signal transducer and activator of transcription 1/2; TYK2, tyrosine kinase 2. Adapted from Oon et al.44
The role of IFN in autoimmunity is highlighted by observations of lupus-like autoimmunity arising de novo in patients receiving treatment with IFN-α for malignancy and chronic viral hepatitis.43 Furthermore, the ISGs are upregulated in several disease states, most notably systemic lupus erythematosus (SLE), where their expression correlates with more severe disease. They are also upregulated in primary Sjögren’s syndrome, systemic sclerosis and a subset of patients with RA.41 Indeed, the potent IFN-I production by pDCs observed following TLR-7/9 ligation by endogenous nucleic acid complexes provides a potential mechanistic basis to explain the provenance of the cytokine in the pathogenesis of SLE, where therapeutic strategies to block the IFN-I pathway are currently in development (see online supplementary Table S1).44
Blocking IFN-α signalling
Sifalimumab is a fully human mAb against multiple IFN-α subtypes, especially IFN-α6, IFN-2b and IFN-2a,45 and has shown promise in a recent phase IIb clinical trial in SLE. In this study,46 431 patients were randomised to receive either monthly infusions of sifalimumab or placebo in addition to standard care. While the trial just missed its primary end point across dosing groups, the highest dose was statistically superior to placebo. As predicted, there was a trend towards a greater effect in patients with a high IFN gene signature.46 Consistent with the role of IFN-α in viral immunity, herpes zoster infections were increased in the sifalimumab group in a dose-dependent manner. Early phase clinical trials of sifalimumab have also been completed in other IMIDs. In dermatomyositis/polymyositis, sifalimumab produced a significant but modest (53%–66%) suppression of a 13-gene IFN signature which positively correlated with clinical improvement in muscle strength.47 However, in a phase I study in psoriasis, sifalimumab failed to suppress the IFN gene signature and had no clinical activity.47
Rontalizumab, a human mAb against all 12 IFN-α subtypes, has been trialled in SLE but failed to reach its primary end point.48 Interestingly, a trend towards efficacy was noted in IFN-low but not in IFN-high patients, and rontalizumab was not associated with increased viral infections.48 These observations may suggest relatively inefficient target engagement, although differences in study design, particularly around management of concomitant immunosuppression, make a direct comparison with sifalimumab difficult. Differences in IFN-α subtype blockade between sifalimumab and rontalizumab may also have influenced their relative efficacy.
Induction of active immunity against IFN-α in a vaccine-based approach is also in clinical development. IFN-α kinoid (IFN-K) is a conjugate protein of inactivated IFN-α coupled to keyhole limpet haemocyanin.49 In a placebo-controlled randomised dose-escalation study of 28 patients with mild-to-moderate SLE, 3 to 4 doses of IFN-K induced anti-INF-α antibodies.49 Interestingly, anti-IFN-α antibody titres were higher in patients with a positive baseline IFN gene signature and correlated negatively with IFN gene expression at day 112.49 The safety of such an approach must await further trials.
In contrast to neutralising IFN-α, it is also possible to block its receptor. Anifrolumab is a human mAb against subunit 1 of the IFN-α receptor (IFNAR) which, in a recent phase IIb clinical trial of 305 patients with SLE, showed efficacy versus placebo both for global and organ-specific disease activity.50 Anifrolumab was also more effective in IFN-high patients and carried a comparable dose-dependent risk of herpes zoster infection to that of sifalimumab.50 In an indirect comparison, anifrolumab appeared to exert a more potent and sustained suppression of IFN gene expression compared with sifalimumab in two Japanese SLE cohorts.51 Anifrolumab is also in phase I development for the treatment of systemic sclerosis.52 53
Upstream inhibition of the IFN-I axis
Several strategies for the upstream inhibition of IFN-I production are currently in early stages of development. BIIB059 is a humanised mAb against BDAC-2, a pDC-specific surface receptor that mediates a reduction in IFN-I production.54 Data from a phase Ib trial in 12 patients with SLE demonstrated a reduction in IFN gene expression and improvement in skin lesions.55 Talacotuzumab is a cytotoxic mAb against CD123, which is expressed in high levels on pDCs.56 Talacotuzumab depleted pDCs from the blood of SLE patients in vitro, leading to inhibition of IFN-I gene expression.56 In addition to antibody-based therapies, a number of small molecular inhibitors of TLR-7/8/9 signalling are currently in development for the treatment of SLE and psoriasis, although results of early phase clinical trials are yet to be formally published.44
Inhibition of type II IFNs
Although type I IFNs are the principal inducers of the IFN gene signature, type II and III IFNs also upregulate these genes.44 IFN-γ signals via a different receptor (IFNGR) than IFN-I, although there is some overlap in downstream signalling cascades40 (figure 2). An mAb against IFN-γ, AMG 811, is in development and has shown dose-dependent reductions in circulating levels of the IFN-γ-dependent protein CXCL1057 and IFN-γ-modulated gene expression in whole blood from patients with SLE.58 Furthermore, treatment with AMG 811 reduced both of these biomarkers in patients with active lupus nephritis in a small (n=28) phase I study, although transiently and with no discernible clinical effect.59 There are currently few data surrounding the role of IFN-λ in autoimmunity, although evidence of a pathogenic role in SLE is emerging.44
Summary: targeting the IFN axis in SLE—what have we learnt?
SLE is a notoriously difficult disease in which to develop novel therapeutics, with many failures and just a single success (belimumab) in the biologic era. While encouraging, the data from targeting of the IFN axis remain early phase and, in part, contradictory. Nonetheless, the biology appears compelling and a multitude of agents are in development. Furthermore, the regulatory approval of Janus knase (JAK) inhibitors provides a further route to directly target IFN signalling, with agents whose pharmacokinetic and pharmacodynamic characteristics are well studied. With this broad armamentarium, and careful trial design, we can look forward to the hypothesis linking IFN activity and SLE to be definitively answered.
Targeting cell adhesion
Integrin blockade
Adhesion molecules play a crucial role in the cell-cell interactions that are necessary for recruitment of circulating immune cells from the vasculature to local tissue sites. Especially important in this regard is the integrin family, which mediates strong adhesion between leucocytes and endothelial and mucosal epithelial cells by binding to extracellular matrix components and specific receptor molecules. Six integrins are expressed only on leucocytes: LFA-1 (αLβ2), Mac-1 (αMβ2), αxβ2, αdβ2, α4β7 and αEβ7.60 Especially notable are: LFA-1, which plays a key role in the formation of the immunological synapse; α4β7, which mediates gut-specific lymphocyte homing via binding to MAdCAM-1 on the surface of gastrointestinal endothelial cells; and αEβ7, which binds E-cadherin on gut epithelial cells and may be important for lymphocyte retention within the mucosa.60 Inhibition of lymphocyte recruitment to end organs can thus be achieved by blocking these interactions, with a specificity determined by the cellular tropism of the target cellular adhesion molecule (figure 3).

Overview of drugs targeted against integrin molecules and their ligands. ICAM-1, intercellular adhesion molecule 1; MAdCAM-1, mucosal vascular addressin cell adhesion molecule 1; VCAM-1, vascular cell adhesion molecule 1. Adapted from Bravatà et al.70
One of the first integrin blockers used in the treatment of autoimmunity was natalizumab, an mAb against the α4 integrin subunit. Natalizumab exerts a relatively non-specific blockade of lymphocyte recruitment at both the blood-brain barrier (α4β1 integrin) and the gut (α4β7 integrin) and is effective in the treatment of multiple sclerosis (MS)61 and CD.62 However, postmarketing surveillance of patients taking natalizumab demonstrated the development of progressive multifocal leukoencephalopathy (PML), a severe and often fatal central nervous system (CNS) infection caused by the JC virus.63 Thus, while natalizumab continues to be used for the treatment of MS, its unfavourable risk:benefit profile limits its use in CD. Although licensed in the USA, it failed to gain regulatory approval for CD in Europe.64 Similarly efalizumab, an mAb against the αL integrin subunit of LFA-1 and effective in the treatment of psoriasis, was withdrawn from the market in 2009 following case reports of PML.65 Nevertheless, topical ocular use of lifitegrast, a small molecule inhibitor of LFA-1, has recently been licensed for the treatment of keratoconjunctivitis sicca.66 Furthermore, two small molecule inhibitors of α4 integrin, carotegrast methyl67 and firategrast,68 are in development for the treatment of ulcerative colitis (UC) and MS, respectively.
In an attempt to reduce the risk of opportunistic infection, more specific integrin inhibitors have been developed. In particular, several mAbs have been developed against the gut-specific α4β7 integrin or its ligand, MAdCAM-1 (see online supplementary Table S2). Furthermore, etrolizumab, an mAb directed solely against the β7 integrin subunit, additionally inhibits binding of αEβ7 to E-cadherin. Whether this translates to superior clinical efficacy is yet to be determined. However, in a phase II study in moderate-to-severe UC, the efficacy of etrolizumab positively correlated with expression of αE in the intestinal mucosa, thereby providing a potential stratification marker for its use.69 To date, these ‘gut-specific’ integrin inhibitors do not appear to be associated with an increased risk of PML.70
Aside from the (brief) use of efalizumab in psoriasis, integrin blockade has so far been of limited clinical utility outside of the setting of CNS and gut autoimmunity. A post hoc analysis of a randomised controlled trial (RCT) of vedolizumab in CD suggested a trend towards resolution of extraintestinal manifestations,71 which was mirrored by preliminary data from a separate cohort of patients with UC and CD.72 However, a case series of new-onset or exacerbated arthritis and sacroiliitis in patients treated with vedolizumab has recently been reported.73 Alongside a bell-shaped dose-response in the UC trial of etrolizumab, these observations may suggest that both pro-inflammatory and anti-inflammatory lymphocyte subsets are targeted by integrin blockade.
Sphingosine-1-phosphate receptor blockade
The sphingosine-1-phosphate (S1P) receptor family comprises five members with effects on cell proliferation; migration and survival; intercellular communication; vascular tone and endothelial barrier function.74 In particular, S1P1 receptor has a key role in the trafficking of lymphocytes out of secondary lymphoid organs. Receptor agonists, via receptor internalisation and degradation, prevent B-cell and T-cell egress into the circulation. Fingolimod, a relatively non-specific small molecule S1P-receptor agonist, is approved for use in relapsing-remitting MS but has been associated with severe herpetic infections, as well as cardiac and hepatic adverse effects.75 Recently, ozanimod, another small molecule agonist but more selective for S1P1 and S1P5 receptors, demonstrated efficacy in a phase II trial in moderate-to-severe UC, with a dose-related reduction in circulating lymphocytes and an acceptable safety profile.76
Summary: targeting cell adhesion — what have we learnt?
Targeting the molecules that underpin immune cell trafficking can have profound effects, both in terms of efficacy but also safety. In particular, the emergence of PML with natalizumab, and perhaps herpetic infection with fingolimod, may indicate a key role in microbiological latency. Nonetheless, an effective and safe therapy may emerge when it is possible to specifically target molecules on key effector subsets, such as the integrins expressed by gut-homing lymphocytes. While long-term safety data are awaited, mucosal αE expression may facilitate the targeting of etrolizumab to patients most likely to benefit from its use.
Janus kinase inhibition
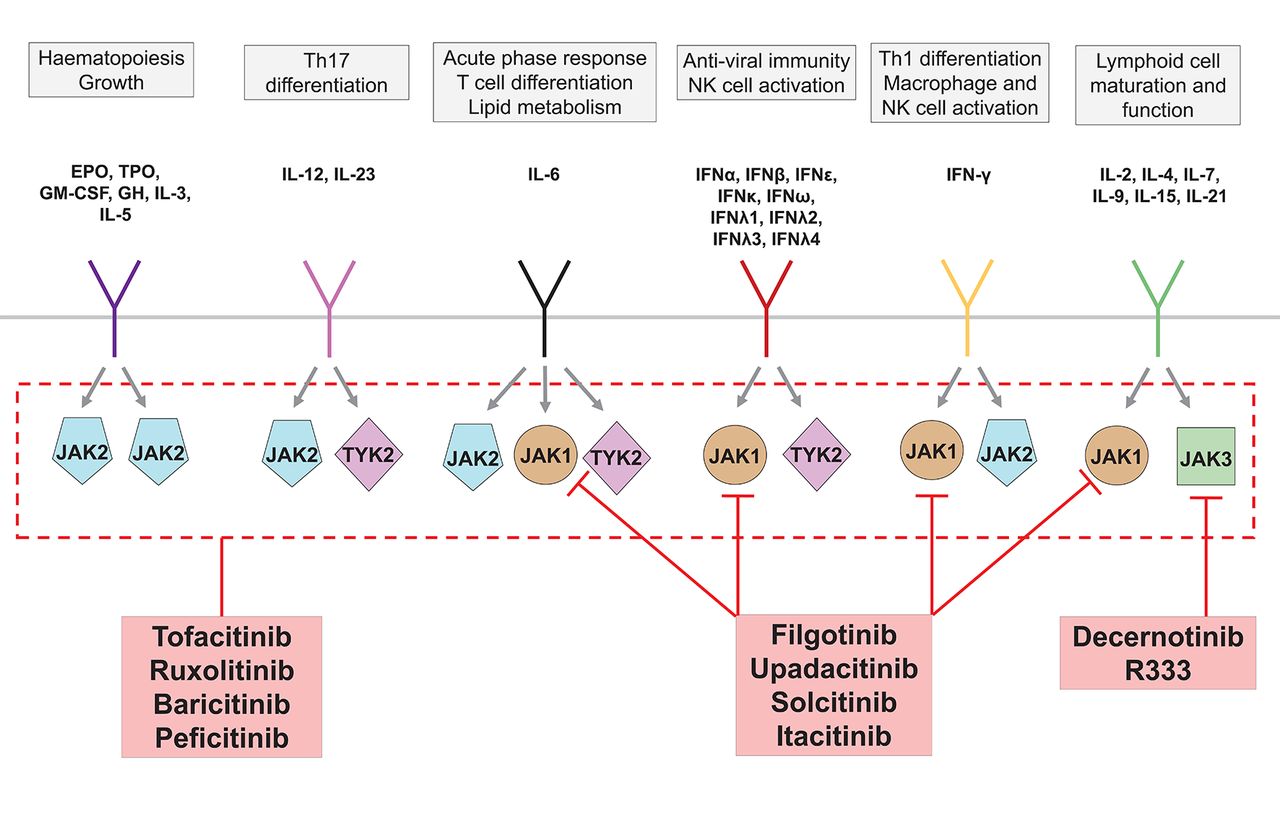
JAKs are intracellular tyrosine kinases that play a crucial role in the signalling pathways of many cytokines involved in immunity and haematopoiesis. On receptor-cytokine binding and receptor dimerisation, receptor-associated JAKs cross-phosphorylate one another. Further phosphorylation of receptor-associated tyrosine residues provides docking sites for STAT proteins, which are also phosphorylated by JAKs.77 Phosphorylated STAT molecules then dimerise and translocate to the nucleus, where they act as potent regulators of gene expression. There are four JAKs—JAK1, JAK2, JAK3 and tyrosine kinase 2 (TYK2)—which function as heterodimers or, in the case of JAK2, also as a homodimer.77 Different JAK dimers associate with different receptors, such that each JAK mediates signalling from a distinct, although overlapping, profile of cytokines (figure 4). Inhibition of JAK signalling therefore offers a novel mechanism by which to block a range of cytokines using a small-molecule drug.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Overview of Janus kinase (JAK) inhibitors developed for the treatment of immune-mediated inflammatory diseases. JAK2-specific inhibitors have been developed for the treatment of haematological malignancy, although are omitted here for simplicity. EPO, erythropoietin; GH, growth hormone; IFN, interferon; GM-CSF, granulocyte macrophage colony-stimulating factor; TPO, thrombopoietin; Tyk2, tyrosine kinase 2. Adapted from reference.77
First-generation JAK inhibitors
Tofacitinib is a pan-JAK inhibitor capable of inhibiting JAK3/1/2 and, to a lesser extent, TYK2.78 Tofacitinib is licensed for its beneficial effects in RA,79 and is currently in development for a range of other IMIDs including juvenile idiopathic arthritis, psoriasis, PsA and UC. Baricitinib and ruxolitinib are JAK1/2 inhibitors which, owing to the heterodimeric functionality of JAKs, exert a very similar spectrum of cytokine blockade to that of tofacitinib and have been trialled in a range of autoimmune diseases (table 2). Ruxolitinib is also licensed for the treatment of myelodysplasia, although the suppressive effects of first-generation JAK inhibitors on haematopoiesis are an unwanted adverse effect in the context of IMIDs. This can be circumvented by topical formulations for dermatological indications,80 although may be an issue for pan-JAK inhibitors when systemic treatment is required.
Development of drugs for IMIDs that target Janus kinases (JAKs). Development status based on http://adisinsight.springer.com, accessed 30 May 2017. AS, ankylosing spondylitis; CD, Crohn’s disease; IMID, immune-mediated inflammatory diseases; JIA, juvenile idiopathic arthritis; P-I, phase I clinical trial; P-II, phase II clinical trial; P-III, phase III clinical trial; SLE, systemic lupus erythematosus; Syk, spleen tyrosine kinase; TYK2, tyrosine kinase 2; UC, ulcerative colitis. *Baricitinib approved by the European Medicines Agency, although rejected by the Food and Drug Administration (April 2017).
Second-generation JAK inhibitors
Recent years have seen the development of a ‘second generation’ of JAK inhibitors for the treatment of IMIDs that exert a selective blockade of JAK1 or JAK3 which, in theory, should have less risk of haematopoietic toxicity—an effect largely secondary to JAK2 inhibition (figure 4).77 However, neutropaenia and lymphopoenia are still encountered in some trials of these agents suggesting either non-redundant JAK1/JAK3/TYK2-dependent haematopoietic mechanisms or, more likely, suboptimal selectivity.81 Indeed, as with most small molecules drugs, target selectivity of JAK inhibitors depends both on the assay used and the concentration/dose studied in vitro or in vivo.81 Combined with further recognised adverse effects including transaminitis, dyslipidaemia, herpes zoster reactivation and lymphopoenia,82 these drugs have a modestly distinct adverse event profile to biologic drugs. Thus, while oral dosing and a rapid onset of efficacy may prove attractive to both practitioners and patients, regulatory approval has not proved straightforward for either tofacitinib83 or baricitinib.84 Furthermore, the focus on JAK1-specific and JAK3-specific inhibition neglects IL-12/IL-23 signalling, which relies on JAK2/TYK2 heterodimers. It is thus unsurprising that JAK1 and JAK3 selective inhibitors have so far proved disappointing in the treatment of psoriasis,85 PsA and AS (table 2). Selective inhibition of TYK2 should have theoretically greater efficacy for these diseases, and several such inhibitors are in early preclinical development.86
Summary: JAK inhibition — what have we learnt?
The development of JAK inhibitors has brought a new approach to the treatment of IMIDs, and rapid onset of biologic-like potency in an oral formulation will prove attractive for diseases such as RA. Furthermore, JAK inhibition has helped to validate aspects of immune physiology, such as the role of γ-chain cytokines in lymphopoiesis. However, JAK inhibitors are of necessity less selective than their biologic counterparts, blocking signalling across multiple cytokine axes simultaneously. Furthermore, as with any small molecule drug, target specificity is not absolute and will depend on the dose delivered to tissues. Consequently, efficacy and toxicity in the clinic may differ from that predicted from in vitro testing, and even from clinical trials. Long-term safety data are therefore required, combined with head-to-head studies, to determine the optimal positioning of JAK inhibitors alongside biologic agents.
Targeting specific cellular subsets
Therapeutic immune modulation can also be achieved via selective depletion, expansion or blockade of specific immune cell subsets. Early examples of this approach include the cell-depleting monoclonal antibodies alemtuzumab (anti-CD52) and rituximab (anti-CD20), which are now licensed for the treatment of MS and RA, respectively. Recent years have seen further development of novel therapies against B-cells as well as agents to expand Tregs, with both therapeutic approaches being trialled in the treatment of IMIDs.
Therapies targeting B cells
Following in the footsteps of rituximab, several other B-cell-depleting mAbs have been developed (see online supplementary Table S3). Ocrelizumab is a humanised anti-CD20 mAb that is effective in the treatment of both relapsing-remitting87 and primary-progressive88 MS. To date, it is the only therapy to demonstrate efficacy in primary progressive MS; rituximab, in comparison, has only proven effective in relapsing-remitting disease.89 It remains to be determined whether this reflects differences in study design, drug posology or a true biological difference between rituximab and ocrelizumab. Nevertheless, development of ocrelizumab in other IMIDs, including RA and SLE, was terminated due to an adverse safety profile, suggesting the possibility of a true difference in the biological function of these two agents despite their common molecular target. Obinutuzumab is an anti-CD20 mAb whose cytotoxic properties have been refined by glycoengineering. B-cell depletion by this afucosylated mAb takes advantage of modified FcγR interactions as well as reduced redistribution and modulation of CD20.90 It is marketed for certain haematological malignancies and is in phase II trials for SLE (NCT02550652).
In addition to cellular depletion, recent years have witnessed the development of several strategies to inhibit B-cell differentiation and survival. B-cell activating factor (BAFF) and APRIL (a proliferation-inducing ligand) are B-cell stimulating molecules important in B-cell maturation and plasma cell survival/class-switching, respectively.91 Attempts to inhibit BAFF and APRIL have, to date, yielded mixed responses. Belimumab, an mAb against soluble BAFF, is marketed for the treatment of SLE, although there is little evidence to support efficacy outside of joint and skin involvement.92 Tabalumab—an mAb against both soluble and membrane-bound BAFF—and the anti-BAFF peptibody blisibimod both exhibited disappointing efficacy for SLE in recent phase III clinical trials.93–95 Both BAFF and APRIL bind to transmembrane activator and calcium-modulator and cyclophilin ligand interactor (TACI).91 Atacicept and RCT 18 are TACI:IgG-Fc fusion peptides capable of blocking both APRIL and BAFF, and both are in development for SLE, although concerns remain surrounding their associated infection risk.91 Perplexingly, BAFF/APRIL antagonism has limited efficacy in RA despite the success of rituximab in this setting. This may in part be explained by B-cell modulatory effects, such as APRIL-mediated IL-10 production by regulatory B cells,96 and the relative functional importance of such mechanisms in different disease settings.
Therapies targeting Treg cells
In contrast to depletion and downregulation of B-cell populations, alternative strategies have been used to stimulate Treg populations to abrogate autoimmunity. One area that has gained recent attention is the use of low-dose recombinant IL-2. Whereas high doses of IL-2 stimulate effector T cells are used in the treatment of certain forms of cancer such as malignant melanoma, low doses preferentially expand Treg populations.97 Indeed, low-dose IL-2 therapy has shown promise in early phase clinical trials in a range of IMIDs including hepatitis C virus-induced vasculitis,98 graft-versus-host disease,99 SLE,100 type I diabetes mellitus101 and alopecia areata.102 Several strategies to boost the tolerogenic effect of low dose IL-2 have been proposed, including the design of ‘second-generation’ IL-2 molecules with longer half-life and improved target cellular profiles, the combination of low-dose IL-2 with existing biological agents and even combination with vaccines to promote antigen-specific tolerance.97
Various alternative approaches to stimulate Tregs using mAbs are in development. TGN-1412 is a super-agonist mAb against the costimulatory molecule CD28. In a notorious phase I study of healthy volunteers in 2006, it caused a life-threatening cytokine storm. This was attributed to CD28-mediated activation of tissue-resident memory T cells—an effect not observed in preclinical cynomolgus macaque studies, in which CD28 is downregulated on these cells.103 Nevertheless, when used at a much lower dose, TGN-1412 can specifically activate Tregs and development has now been relaunched under the name theraliximab.104 Tregalizumab is an mAb against CD4 and, in contrast to other CD4 mAbs, binds to a distant epitope and has been shown to specifically activate Tregs in preclinical studies.105 Nevertheless, a phase IIb study in RA failed to show improvement in ACR20 response compared with placebo,105 and further development of the drug for this indication has been discontinued.
Recent years have seen growing interest in so-called Treg epitopes (Tregitopes)—highly conserved amino acid sequences within IgG molecules which can be presented on a wide range of major histocompatibility complex-II alleles to selectively activate Tregs.106 It has been proposed that Tregitopes represent an evolutionary mechanism by which to suppress autoreactivity to the wide array of different immunoglobulin molecules that are created during immune development,107 and may be the mechanism underlying the efficacy of intravenous immunoglobulin in the treatment of IMIDs.108 Tregitopes can ameliorate inflammation in several murine models of autoimmunity, and are in preclinical stages of development for the treatment of IMIDs.107
Several cellular therapeutic approaches to enhance Treg function are also in the early stages of development for the treatment of IMIDs, including exogenous Treg transfer109 and tolerogenic dendritic cell therapies.110 111
Summary: targeting specific cellular subsets — what have we learnt?
Recent years have witnessed a rapid expansion in the array of biologic therapies to selectively deplete or inhibit B-cells, and the emergence of therapeutic strategies aimed at expanding Tregs. Novel aspects include glycoengineering to optimise depleting potency, where required. However, despite apparent success in preclinical development, efficacy in later stage clinical trials has been somewhat mixed. For B-cell targeted therapy this may reflect the various B-cell subsets and their heterogeneous function(s). Thus, more focused therapy may be required to optimise efficacy. Posology of these agents is also clearly of importance, as demonstrated by the widely contrasting effects of low -dose and high-dose IL-2 and TGN-1412 therapies. In terms of cellular therapies, the long-term stability of therapeutically expanded Tregs and the risk of conversion to an effector phenotype remains uncertain.112 Furthermore, antigen-specific approaches are likely to provide the optimal route for cell-targeted therapies.
Newer technologies
Bispecific antibodies
Despite the potent blockade of cytokine signalling afforded by biologic therapies, many patients have only a partial or transient response. In some cases, this is attributable to immunogenicity against the biologic agent, although in other cases likely reflects redundancy and/or plasticity in the underlying autoimmune processes. Attempts to block multiple cytokines through the simultaneous use of different biologics have, however, been limited by unacceptable adverse effects without superior efficacy.113–115
With advances in mAb technology, a number of approaches enable the targeting of multiple molecular species by a single therapeutic.116 For example, ABT-122 is a so-called dual variable domain mAb against both IL-17 and TNF-α. In small, early phase studies it appears to have a similar safety profile to adalimumab in PsA and RA.117 Furthermore, in a phase II trial in patients with PsA with an inadequate response to methotrexate, there was some evidence of superiority of ABT-122 when compared with adalimumab for both ACR70 (ABT-122 vs ADA, 31.5% vs 15.3%, p<0.05) and PASI75 (77.6% vs 57.6%, p<0.05) responses.118 In contrast, a phase Ib/IIa trial in psoriasis of COVA322, a so-called fynomab that targets both IL-17 and TNF-α, was terminated due to safety concerns (NCT02243787, results not published).
The original trials combining two biologic drugs can be criticised for not studying sufficiently low doses of these potentially synergistic combinations. A disadvantage of bispecific and trispecific reagents, however, is that they only allow fixed ratios of cytokine blockade to be tested.
Gene therapy approaches
Mongersen is a modified release antisense nucleotide to mothers against decapentaplegic homolog 7 (SMAD-7), designed to be released into the terminal ileum and proximal colon.119 SMAD-7 is central to transforming growth factor-β1 signalling, itself important in the pathogenesis of CD.120 In a phase II trial, a short course of mongersen proved superior to placebo at inducing remission in patients with active CD.121 Most adverse events were attributable to the disease itself, and this trial provides proof of principle that it is possible to interfere with immunopathological processes at the level of gene transcription, by local delivery of a nucleotide-based therapy.
Summary — what have we learnt from newer technologies?
It is early days but the first trials of bispecific antibodies have provided mixed results. A raft of agents are in development, including trivalent nanobodies and PEGylated single chain fragment variables.122 Not all of these drugs will reach the clinic, certainly in IMIDs. Furthermore, in some cases, the anticipated advantages of poly-targeting could be offset by lack of effector function and short half-lives, but careful choice of disease and trial design should mitigate against these potential shortcomings. In terms of gene therapy, it is again early days but there is clearly the potential for local delivery of such agents in articular diseases, as in CD.
Conclusions
The future remains exciting for clinicians treating IMIDs, and for their patients. Targeted therapies, as well as providing new treatment paradigms, continue to inform us about the pathogenesis of disease and its complications. In this brief review, we have highlighted just a few examples of novel approaches, particularly where data have provided new downstream knowledge and teachings. However, many challenges remain—in particular, the ability to target these various approaches to both the diseases and the patients who are most likely to benefit. Equally challenging is the need for head-to-head comparisons between different agents, to reliably dissect the relative contributions of distinct pathways to a particular disease. Future trials will need to become increasingly sophisticated in order to address these varying requirements.
References
Footnotes
Handling editor Tore K Kvien
Contributors Conception: JDI. Design, analysis, drafting and final approval of the manuscript: JDI, KFB.
Funding KFB is supported by a Wellcome Trust Fellowship in Translational Medicine and Therapeutics (102595/Z/13/A) and a grant from the National Institute for Health Research Newcastle Biomedical Research Centre (BH136167/PD0045) based at Newcastle Hospitals NHS Foundation Trust and Newcastle University. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Competing interests JDI has received honoraria, research grants or consulting fees from AbbVie; Biogen; Bristol-Myers-Squibb Company; Celltrion Healthcare; Chugai Pharmaceutical; Eli Lilly and Company; Hospira; Janssen Pharmaceuticals; Merck Serono S A; Pfizer; and Roche Laboratories. KFB has no competing interests to declare.
Provenance and peer review Commissioned; externally peer reviewed.
Data sharing statement None declared.