Article Text

Abstract
Objective Interleukin (IL)-12 and IL-23 have been implicated in the pathogenesis of rheumatoid arthritis (RA). The safety and efficacy of ustekinumab, a human monoclonal anti-IL-12/23 p40 antibody, and guselkumab, a human monoclonal anti-IL-23 antibody, were evaluated in adults with active RA despite methotrexate (MTX) therapy.
Methods Patients were randomly assigned (1:1:1:1:1) to receive placebo at weeks 0, 4 and every 8 weeks (n=55), ustekinumab 90 mg at weeks 0, 4 and every 8 weeks (n=55), ustekinumab 90 mg at weeks 0, 4 and every 12 weeks (n=55), guselkumab 50 mg at weeks 0, 4 and every 8 weeks (n=55), or guselkumab 200 mg at weeks 0, 4 and every 8 weeks (n=54) through week 28; all patients continued a stable dose of MTX (10–25 mg/week). The primary end point was the proportion of patients with at least a 20% improvement in the American College of Rheumatology criteria (ACR 20) at week 28. Safety was monitored through week 48.
Results At week 28, there were no statistically significant differences in the proportions of patients achieving an ACR 20 response between the combined ustekinumab group (53.6%) or the combined guselkumab group (41.3%) compared with placebo (40.0%) (p=0.101 and p=0.877, respectively). Through week 48, the proportions of patients with at least one adverse event (AE) were comparable among the treatment groups. Infections were the most common type of AE.
Conclusions Treatment with ustekinumab or guselkumab did not significantly reduce the signs and symptoms of RA. No new safety findings were observed with either treatment.
Trial registration number NCT01645280.
- Rheumatoid Arthritis
- Treatment
- Disease Activity
- Methotrexate
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
For patients with moderate-to-severe rheumatoid arthritis (RA), treatment with biologic tumour necrosis factor (TNF) inhibitors and other targeted therapies with different modes of action is often effective in reducing joint symptoms and inhibiting progression of damage.1–7 However, many patients with RA do not respond to or lose response over time to the currently available treatments,8 ,9 thus, there remains a need for novel therapies.
Interleukin (IL)-12 upregulates T helper type 1 (Th1) cell differentiation, and is the main stimulator of interferon (IFN)-γ. Increased levels of IL-12 have been detected in serum and synovial fluid of patients with RA and correlate with disease activity.10 Also, IFN-γ is upregulated in RA synovial membranes,11 and an IFN-γ signature can be found in peripheral mononuclear cells of patients with RA.12 IL-23 is a member of the IL-12 cytokine family and activates Th17 cells leading to increased production of several other cytokines, including IL-17 and TNF. Various in vivo models have demonstrated that the IL-23-Th17 pathway may be involved in the development of autoimmunity, including RA.13–15 Elevated levels of IL-23 have been observed in serum and synovial fluid of patients with RA, and serum levels decreased following anti-TNF therapy.16–19 Furthermore, Th17 cell numbers as well as synovial IL-17 expression were found to be increased in RA.20 ,21 Importantly, the extent to which RA is governed by Th1 or Th17 cells remains unclear. Therapies targeting the Th1 pathway have not been evaluated in RA, and therapies targeting IL-17 have shown variable efficacy in RA.22
Ustekinumab is a human monoclonal antibody targeting the IL-12/23 p40 subunit, therefore inhibiting both IL-12 and IL-23 activities, and guselkumab is an investigational monoclonal antibody targeting IL-23 specifically. Ustekinumab is effective in treating moderate-to-severe psoriasis23 ,24 and active psoriatic arthritis (PsA), including inhibition of radiographic progression through 2 years.25 ,26 Results of a phase II trial suggest that guselkumab may be effective in treating psoriasis;27 guselkumab is currently being studied in phase III trials in psoriasis and a phase II trial in PsA. The current phase II trial was conducted to evaluate the efficacy and safety of ustekinumab and guselkumab in patients with active RA despite concomitant methotrexate (MTX).
Patients and methods
Patients
Adults (18–80 years) with a diagnosis of RA, according to the American College of Rheumatology (ACR) 1987 criteria,28 for ≥6 months with persistent disease activity despite treatment with MTX were eligible. Persistent disease activity was defined as: ≥6/66 swollen joints and ≥6/68 tender joints and a serum C reactive protein (CRP) level ≥0.8 mg/dL. Eligibility criteria included a positive test for anticyclic citrullinated peptide antibodies or rheumatoid factor and prespecified tuberculosis screening. Patients who had received any approved or investigational biologic agent were not eligible.
Study oversight
This trial was conducted in accordance with the principles of the Declaration of Helsinki. Each patient gave written informed consent. This study was sponsored by Janssen Research & Development, LLC. The authors, some of whom are employees of the study sponsor, participated in designing the study and collecting and analysing the data. An independent data monitoring committee regularly reviewed unblinded interim safety data. All authors drafted or revised the manuscript with the assistance of a professional medical writer employed by Janssen Scientific Affairs, LLC.
Study design
This was a phase II, randomised, double-blind, placebo-controlled, parallel group trial. All patients had received MTX (10–25 mg per week) for ≥6 months before screening, and the dose was to be stable for ≥12 weeks prior to randomisation. Stable doses of concomitant glucocorticoids (<10 mg prednisone/day) and/or non-steroidal anti-inflammatory drugs (NSAIDs) and/or other analgesics for RA were permitted. Patients were randomly assigned (1:1:1:1:1) to receive placebo at weeks 0, 4 and every 8 weeks, ustekinumab 90 mg at weeks 0, 4 and every 8 weeks, ustekinumab 90 mg at weeks 0, 4 and every 12 weeks, guselkumab 50 mg at weeks 0, 4 and every 8 weeks, or guselkumab 200 mg at weeks 0, 4 and every 8 weeks through week 28. To maintain the blind, all randomised patients were to receive two 1 mL subcutaneous injections in two identical syringes prepared by an independent unblinded pharmacist at the sites at weeks 0, 4, 12, 16, 20 and 28 according to treatment assignment, including placebo injections as needed to maintain the blind, depending on the dose of ustekinumab or guselkumab assigned. The doses chosen for ustekinumab and guselkumab were based on the efficacy seen with the respective doses in previous trials of PsA and/or psoriasis.23–27 At week 16, patients in the placebo group who had <10% improvement from baseline in both tender and swollen joint counts entered double-blind early escape and received ustekinumab 90 mg at weeks 16, 20 and 28; no treatment adjustments were made for patients randomised to the ustekinumab or guselkumab groups. The final safety follow-up visit was at week 48.
Efficacy
The primary end point was the proportion of patients who achieved at least a 20% improvement in the ACR criteria (ACR20)29 at week 28. Other efficacy assessments included the 28-joint count Disease Activity Score using CRP (DAS28-CRP),30 Clinical Disease Activity Index (CDAI) and Simplified Disease Activity Index (SDAI).31 Physical function and health-related quality of life were assessed using the Health Assessment Questionnaire-Disability Index (HAQ-DI) and the 36-item Short-Form Health Survey (SF-36), respectively.32 ,33
Safety
Patients were monitored through week 48 for adverse events (AEs), clinical laboratory testing, vital signs and ECGs (weeks 0, 16 and 28).
Pharmacokinetics and immunogenicity assessments
Blood samples were collected for measuring serum ustekinumab and guselkumab concentrations and for evaluation of antidrug antibodies. Serum guselkumab concentrations were measured using a validated dissociation-enhanced lanthanide fluorescent immunoassay (lowest quantifiable guselkumab concentration: 0.04 μg/mL). Serum ustekinumab concentrations were measured by a validated electrochemiluminescence-based immunoassay method (lowest quantifiable ustekinumab concentration: 0.17 μg/mL). The presence of antibodies to ustekinumab or guselkumab in serum was determined using validated electrochemiluminescence immunoassays.
Statistical analysis
The study was powered to detect a difference in the proportion of patients achieving an ACR20 response at week 28 in the combined ustekinumab group and the combined guselkumab group compared with placebo. Combining the two dose groups of ustekinumab and guselkumab was prespecified to increase the power for detecting a difference between either ustekinumab or guselkumab and placebo as both dose groups were expected to be efficacious based on pharmacokinetic and efficacy data from other indications.23–27 Based on a simulation of 5000 repetitions, a sample size of 50 patients per treatment group was predicted to provide approximately 74–89% power to detect an approximately 20–30% difference between a placebo plus MTX group and an active treatment (ustekinumab or guselkumab) plus MTX group. To control an overall type I error at 0.05 through the trial, a two-sided Cochran-Mantel-Haenszel test with stratification by screening CRP level (<1.5, ≥1.5 mg/dL) at a significance level of 0.025 was used in each step of a sequential testing process. For both ustekinumab and guselkumab, the combined group was compared with placebo first; if the difference from placebo was significant, pairwise comparisons between each dose group and placebo were performed. The primary end point analysis included all randomised patients grouped by randomised treatment. If a patient had data for at least one ACR component at week 28, missing component data were imputed with the last observation carried forward if baseline data were available; otherwise, missing components were considered to contribute to less than 20% improvement for ACR20 response. Patients were classified as non-responders if no ACR component data were available at week 28 or if they initiated prohibited medications (including glucocorticoids for RA), increased the MTX or glucocorticoid dose above baseline level, or discontinued the study agent for any reason. For patients who entered early escape, week-16 efficacy values were carried forward through week 28. A nominal significance level of 0.05 (two-sided) was applied to secondary end points and other analyses.
Results
Patients
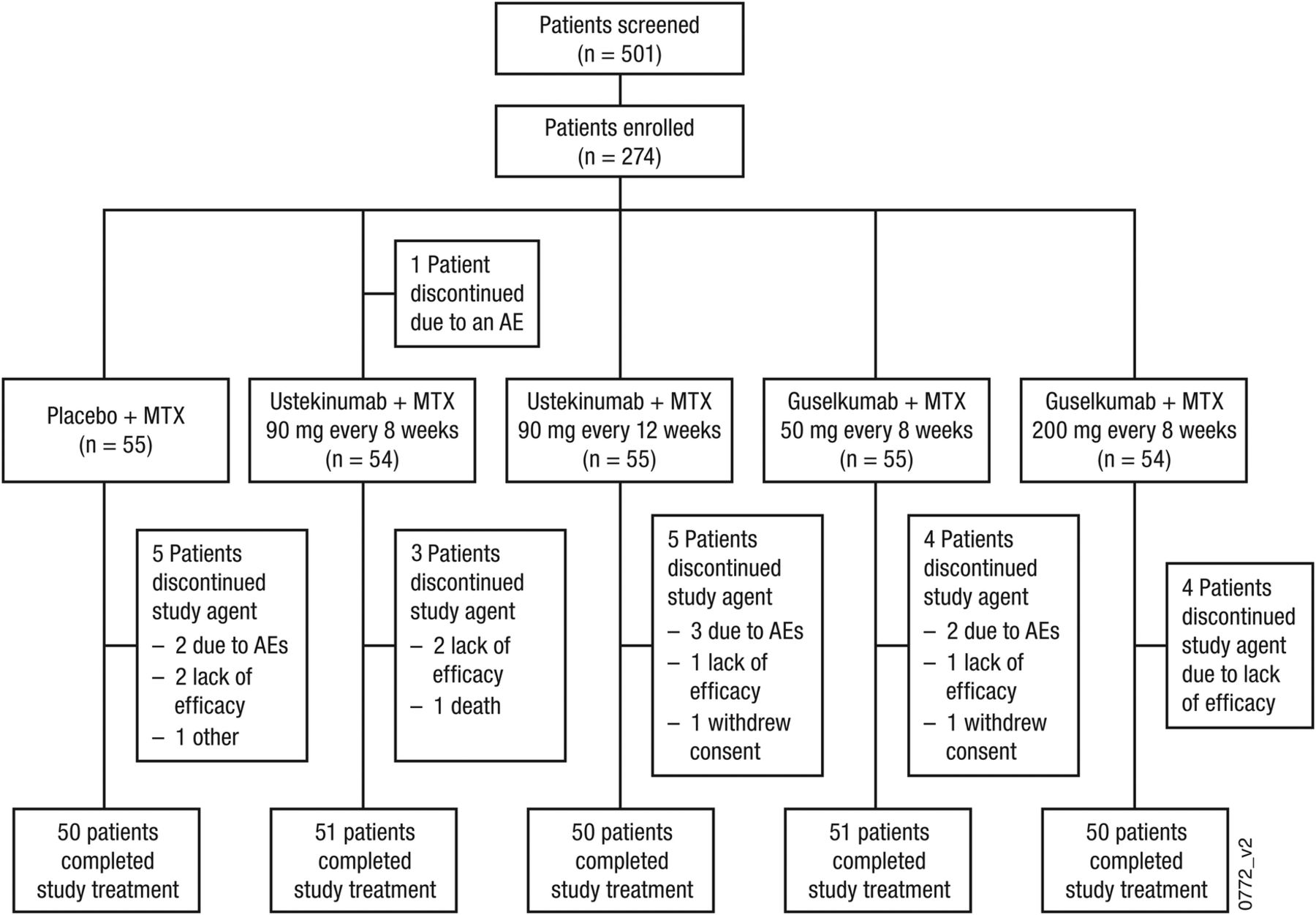
Data were collected from July 2012 to May 2014 at 59 sites in the USA (n=1), South America (n=72), Europe (n=197) and Asia (n=4). Five hundred and one patients were screened and 274 were randomised (placebo plus MTX, n=55; ustekinumab 90 mg every 8 weeks plus MTX, n=55; ustekinumab 90 mg every 12 weeks plus MTX, n=55; guselkumab 50 mg plus MTX, n=55; guselkumab 200 mg plus MTX, n=54) (figure 1). Baseline demographics and disease characteristics were similar across treatment groups (table 1 and online supplementary table S1). Through week 28, 22 patients discontinued the study agent; the most common reasons were lack of efficacy (n=10, 3.6%) and AEs (n=8, 2.9%) (figure 1).
Baseline demographic and disease characteristics

Patient disposition through week 28. AE, adverse event; MTX, methotrexate.
supplementary tables
Efficacy
The primary end point was not achieved; all significant differences in secondary end points are considered nominal. At week 28, an ACR20 response was achieved by 40.0% of patients in the placebo group, 53.6% in the combined ustekinumab group and 41.3% in the combined guselkumab group (p=0.101 and p=0.877, respectively); ACR20 responses in each dose group are shown in figure 2. Compared with placebo, no treatment benefit on ACR20 response was observed with ustekinumab or guselkumab in any subgroup defined by baseline demographics, disease characteristics or concomitant medications (MTX, oral glucocorticoids or NSAIDs) when compared with placebo (see online supplementary figures S1–S12). However, among ustekinumab-treated patients, a numerically greater proportion of patients in Europe achieved an ACR20 response over placebo compared with those in South America, where a high placebo response rate (58.3%) was observed. Per cent improvements in the majority of ACR components, most notably tender and swollen joint counts and physician's global assessment of disease activity, were numerically greater in the combined ustekinumab group compared with placebo at week 28; however, the effect on CRP was similar to that observed with placebo (table 2 and online supplementary table S2). No consistent improvements in ACR components were observed in the combined guselkumab group except for modest decreases in swollen and tender joint counts and physician's global assessment of disease activity, particularly in the 200 mg group.
Secondary efficacy assessments at week 28
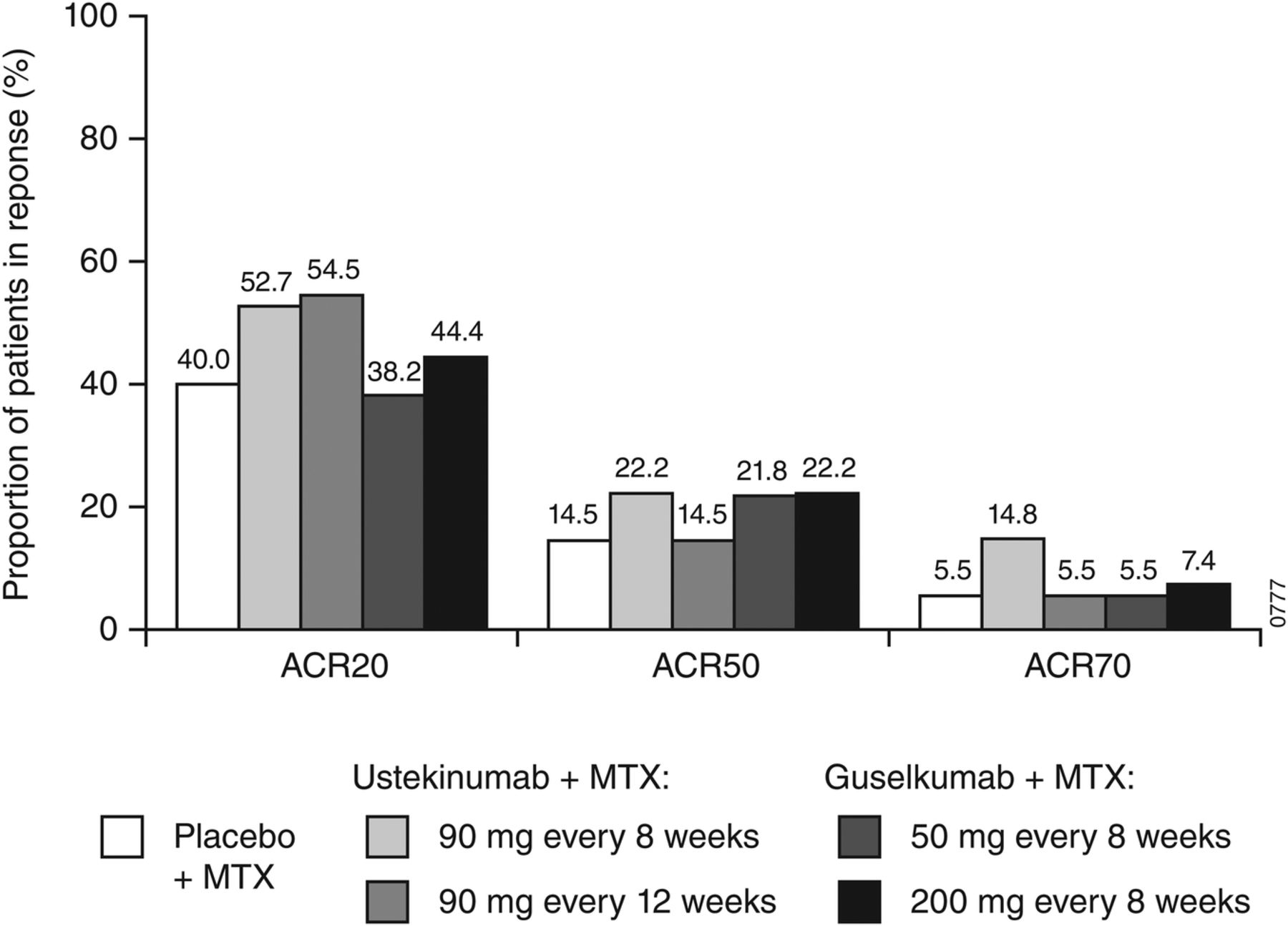
Proportions of patients with an ACR20, ACR50 or ACR70 response at week 28. ACR20 response includes all randomised patients. ACR50 and ACR70 responses include patients who received ≥1 dose of study agent. ACR20/50/70, ≥20%/50%/70% improvement in the American College of Rheumatology criteria; MTX, methotrexate.
supplementary tables
Greater improvements from baseline in DAS28-CRP at weeks 12 and 28 were observed in the combined ustekinumab group but not in the combined guselkumab group compared with placebo at both weeks 12 and 28 (nominal p<0.05) (table 2 and online supplementary table S2). Mean changes from baseline in CDAI and SDAI were greater in the combined ustekinumab group compared with placebo at weeks 12 and 28 and in the combined guselkumab group compared with placebo at week 28 (nominal p<0.05) (table 2 and online supplementary table S2), with a trend of improvement in CDAI and SDAI Scores over time (figure 3). No improvements in mean HAQ-DI Scores were observed in both the combined ustekinumab and combined guselkumab groups at weeks 12 and 28 (table 2 and online supplementary table S2).

{kind=link}
{kind=link}
{kind=link}
Mean Clinical Disease Activity Index (CDAI; panel A) and Simplified Disease Activity Index (SDAI; panel B) Scores through week 28. MTX, methotrexate.
Safety
Through week 16, before early escape, and through week 48, the proportions of patients with at least one AE were generally comparable among the treatment groups (table 3). Through week 48, infections were the most common type of AE. Four patients had a serious infection (placebo: appendicitis (n=1); ustekinumab 90 mg every 8 weeks: urinary tract infection (n=1); guselkumab 200 mg: lobar pneumonia and gastroenteritis (n=1 each)), with no apparent difference among the treatment groups. There were no cases of tuberculosis or opportunistic infections. Overall, gastrointestinal AEs were more common in the ustekinumab 90 mg every-12-week group and blood and lymphatic disorder AEs were more common in the guselkumab 200 mg group compared with placebo (5 (9.1%) vs 2 (3.6%) and 5 (9.3%) vs 1 (1.8%), respectively). Few patients had an injection site reaction (table 3); none were serious or severe, and none led to discontinuation of the study agent.
Adverse events summary through week 48
The incidence of patients with at least one serious AE (SAE) through week 48 was low and similar across treatment groups (table 3). Two malignancies occurred, both during the follow-up period after week 28: squamous cell carcinoma of the lung (male, aged 59 years, non-smoker) in the ustekinumab 90 mg every-12-week group and breast cancer (female, aged 62 years) in the guselkumab 200 mg group. One death occurred in a woman aged 61 years in the ustekinumab 90 mg every-8-week group who experienced syncope (16 days after receiving the third administration of study drug) and was admitted to the hospital, with pulmonary embolism or pneumonia considered at admission; the exact cause of death could not be confirmed. Other SAEs were unstable angina and worsening of RA (placebo); sciatica, anaemia, concussion and shock (ustekinumab 90 mg every 8 weeks); and ileus and transient ischaemic attack (ustekinumab 90 mg every 12 weeks). All SAEs were singular events, and no specific pattern of association between SAEs and active treatments was identified.
There were no clinically meaningful changes in vital signs or ECG findings. Most chemistry and haematology abnormalities were mild to moderate; occurrences of toxicity grades >2 were transient and not clinically significant.
Pharmacokinetics
Median trough serum levels of ustekinumab reached steady state by week 12 (1.59 μg/mL) in the every-8-week group and by week 16 (0.61 μg/mL) in the every-12-week group, and were maintained through week 28 (1.77 µg/mL and 0.54 µg/mL, respectively). No clear correlation between trough serum ustekinumab concentrations and ACR20 response at week 28 was observed.
Median trough serum levels of guselkumab reached steady state by week 20 and were maintained through week 28 (0.18 µg/mL and 0.73 μg/mL in the 50 mg every-8-week and 200 mg every-8-week groups, respectively).
Immunogenicity
Through week 48, 7 out of 123 (5.7%) ustekinumab-treated patients with appropriate samples tested positive for antibodies to ustekinumab; four had neutralising antibodies. In both ustekinumab groups, serum ustekinumab concentrations were generally lower in patients who tested positive for antibodies to ustekinumab compared with those who tested negative.
Through week 48, 12 out of 106 (11.3%) guselkumab-treated patients with appropriate samples tested positive for antibodies to guselkumab; none had neutralising antibodies. Serum guselkumab concentrations were generally comparable between patients who tested positive for antibodies to guselkumab and those who tested negative.
Discussion
The relative contributions of IL-12 and/or IL-23 pathways to the pathophysiology of RA are not well understood. This trial was undertaken to evaluate the safety and efficacy of ustekinumab (anti IL-12/23p40 antibody) and guselkumab (anti-IL-23 antibody) in patients with active RA despite MTX therapy. The primary end point (ACR20 at week 28) was not met for either ustekinumab or guselkumab. While some numerical trends towards improvement were consistently observed in several secondary efficacy measures in the ustekinumab treatment groups compared with the placebo, the reductions in composite disease activity measures DAS28-CRP, SDAI and CDAI were relatively small. No consistent evidence of efficacy in RA was observed with guselkumab in this study. No treatment effect was observed with ustekinumab or guselkumab on CRP levels in patients with RA.
The large placebo effect on ACR20 response observed at week 12 (29.1%) and week 28 (40.0%) may have made it more difficult to demonstrate efficacy for the active treatments, which may be a limitation for this study. However, it should be noted that placebo response rates have been quite high in several recent trials of similar populations, such as REALISTIC (ACR20 response: 26% at week 1234) or MOBILITY (ACR20 response: 46% at week 12 and 33% at week 2435 ,36); however, while the placebo rates were in the order of those observed in our trial, the response rates with the active medications were in the order of 51% to 72% and thus much higher and even up to double those observed here. The small sample size in this phase II study could also be a limitation. However, the totality of the data, including ACR 20/50/70 response over time and lack of effect on CRP, an objective measure, suggested ustekinumab and guselkumab did not have significant, clinically meaningful effects on the signs and symptoms and the inflammatory markers of RA in this patient population with moderate-to-severe disease.
As both guselkumab (which selectively inhibits IL-23) and ustekinumab (which blocks IL-12/IL-23) failed to demonstrate robust efficacy in this study, these results suggest that activation of Th17 cells may not play a major role in established RA. These data are in stark contrast to those obtained in psoriasis with both monoclonal antibodies and in PsA with ustekinumab. Indeed, the efficacy of ustekinumab in psoriasis23 ,24 and PsA,25 ,26 and of guselkumab in psoriasis27 is consistent with the effects of IL-17 inhibition in both diseases. Notably, the lack of robust efficacy in patients with RA following treatment with guselkumab or ustekinumab in the present trial is also consistent with previous studies on IL-17 inhibition in patients with moderate-to-severe RA, which showed modest efficacy with secukinumab37 and ixekizumab38 and no efficacy with brodalumab.39 Overall, these findings point to differences in the immunopathology of active RA and PsA joint disease; in PsA, the IL-23/IL-17-mediated Th17 pathway may play a more important role.
While the lack of demonstrable efficacy for both ustekinumab and guselkumab in patients with RA suggests that Th17 cells may only play a minor role in established RA, the effect of ustekinumab on Th1 cells suggests that Th1 cells play a limited role in the pathophysiology of RA at this (established) stage of the disease. Alternatively, Th1 cell function may be blocked insufficiently at the ustekinumab dose level evaluated in this study. However, in a previous study, evidence of inhibition of IFNγ production was shown in a subset of patients with psoriasis who had ≥75% improvement in Psoriasis Area and Severity Index after receiving a single administration of ustekinumab.40
Interestingly, abatacept, a T cell activation inhibitor, has shown robust efficacy on joint symptoms in both RA41 and PsA,42 while an effect on skin psoriasis was not observed in a phase II PsA Study.42 However, with the exception of abatacept, no T cell directed therapy has hitherto shown efficacy in RA.43 Also, it is not clear at present if the major mode of action of abatacept in RA is mainly related to inhibition of T cell activation or due to other mechanisms.44–46 Irrespective of abatacept's mode of action, the difference in efficacy profiles of ustekinumab, guselkumab and abatacept as well as direct IL-17 inhibitors in RA,37 ,39 ,47 PsA48 ,49 and psoriasis50 ,51 suggest that the IL-23/IL-17-mediated Th17 pathways do not play an important role in RA joint inflammation.
An alternative explanation could be that the highest ustekinumab and guselkumab exposures achieved in this study may not have been adequate for efficacy in RA. However, this is unlikely, as the serum exposures of ustekinumab and guselkumab observed in this study were generally consistent with those observed in different patient populations (eg, psoriasis and PsA) that demonstrated efficacy. Furthermore, no consistent dose response in efficacy was observed for either ustekinumab or guselkumab, and there was no clear correlation between steady state trough serum concentrations of ustekinumab or guselkumab with the proportion of patients who achieved an ACR20 response.
The safety profiles of ustekinumab and guselkumab were consistent with earlier studies in other patient populations.23–27 Overall, the incidence of AEs in the ustekinumab and guselkumab groups was comparable with the placebo group through week 16. The incidence of SAEs was low and was similar among the treatment groups, with no specific pattern of association between SAEs and active treatments. Four patients (one receiving placebo, one receiving ustekinumab, two receiving guselkumab) had a serious infection. No opportunistic infections or cases of tuberculosis were reported. Two malignancies (one in a patient who received ustekinumab and one in a patient who received guselkumab) and one death occurred (ustekinumab group). Overall, no new safety risk or particular pattern of event clustering was evident.
In summary, patients with active RA despite prior MTX did not demonstrate any clinically meaningful improvement in the signs and symptoms of RA following treatment with ustekinumab or guselkumab despite the clear benefit of ustekinumab in both psoriasis and PsA and robust phase II data suggesting efficacy of guselkumab in psoriasis. Our results suggest that, in contrast to psoriasis and PsA, Th17 cells, as well as Th1 cells, may not play a major role in the pathogenesis of active established RA. Additional research is needed to fully understand the relative roles of IL-12 and IL-23 in RA.
Acknowledgments
The authors thank Mittie Doyle, MD, and Alan Mendelsohn, MD, formerly of Janssen Research & Development, LLC, and Yasmine Wasfi, MD, Surekha Mudivarthy, PhD, and Weichun Xu, PhD, of Janssen Research & Development, LLC, for their contributions to this study. The authors also thank Rebecca E Clemente, PhD, of Janssen Scientific Affairs, LLC, for writing support.
References
Footnotes
Handling editor Tore K Kvien
Investigators: Argentina: Horacio Oscar Venarotti, Buenos Aires; Rodolfo Ariel Pardo Hidalgo, San Juan; Guillermo Tate, Buenos Aires; Alberto Spindler, Tucumán; María Alicia Lázaro, Buenos Aires. Bulgaria: Zlatimir Kolarov, Sofia; Boycho Oparanov, Sofia; Anastas Batalov, Plovdiv. Chile: Sonia Arriagada, Osorno. Colombia: John Londono, Chia; William Otero, Bucaramanga; Jose Fernando Molina, Medellin; Patricia Velez, Bogota; Maria Claudia Diaz, Bogota; Diego Luis Saaibi, Bucaramanga; Beatriz Arana, Cali; Luis Fernando Pinto, Medellin. Czech Republic: Gabriela Simkova, Kladno; Libor Novosad, Hlucin; Eva Dokoupilova. Hungary: Edit Drescher, Veszprém; Bernadette Rojkovich, Budapest; Edit Agnes Toth, Gödöllő, Lilla Nafradi, Szombathely; Kata Kerekes, Szekesfehervar. Poland: Piotr Lesacaynski, Poznan; Artur Racewicz, Bialystok; Anna Zubrzycka-Sienkiewicz, Warszawa; Przemyslaw Kotyla, Sosnowiec; Mariusz Korkosz, Krakow; Malgorzata Sochocka-Bykowska, Sopot; Maria Rell-Bakalarska, Warszawa; Elzbieta Langer-Bieda, Krakow; Jan Brzezicki, Elblag. Russia: Marina Stanislav, Moscow; Alexey Maslyanskiy, St. Petersburg; Irina Marusenko, Petrozavodsk; Natalia Shilkina,Yaroslavl; Yurii Shvartz, Saratov; Rimma Kamalova, Ufa; Andrey Rebrov, Saratov; Leysan Myasoutova, Kazan; Elena Zonova, Novosibirsk; Irina Vinogradova, Ulyanovsk. Singapore: Tang Ching Lau, Novena; Kam Hon Yoon, Boon Keng. Ukraine: Oleksander Dyadyk, Donetsk; Andriy Gnylorybov, Donetsk; Volodymyr Kovalenko, Kiev; Andriy Petrov, Simferopol; Mykola Stanislavchuk, Vinnitsa; Roman Yatsyshyn, Ivano-Frankovsk; Svitlana Smiyan, Ternopil; Vira Tseluyko, Kharkiv; Samvel Turianytsia, Uzhgorod. USA: John Budd, St. Louis, Missouri; Mitchell Lowenstein, Palm Harbor, Florida; Michael Miniter, Rock Island, Illinois.
Contributors Study design: SKA, EI, XLX, WR, AG, AB, DB; Data collection, analysis and/or interpretation; drafting or revising the manuscript; approval to submit manuscript: JSS, SKA, EI, XLX, YM, YZ, IN, WR, AG, AB, DB.
Funding This study was funded by Janssen Research & Development, LLC.
Competing interests JSS has received grants from AbbVie, BMS, Janssen, Lilly, MSD, Pfizer and Roche and has served as a consultant for AbbVie, Amgen, Astra-Zeneca, Astro, BMS, GlaxoSmithKline, Janssen, Lilly, MSD, Novartis, Pfizer, Roche, Samsung, Sanofi-Aventis and UCB. SKA served as a Steering Committee member for Janssen. EI served as a trial investigator for Janssen. XLX, YZ, IN, AG, and DB and YM are employees of Janssen Research & Development, LLC, and own stock in Johnson & Johnson, of which Janssen Research & Development, LLC, is a wholly owned subsidiary. AB and WR were employees of Janssen Research & Development, LLC, at the time this work was performed and own stock in Johnson & Johnson, of which Janssen Research & Development, LLC, is a wholly owned subsidiary. WR is currently employed at Sandoz, Inc., Princeton, NJ.
Ethics approval Institutional review board or ethics committee at each site.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Unpublished data from this trial are not currently publicly available.