Article Text

Abstract
Objectives Determine the efficacy and safety of daily lesinurad (200 or 400 mg orally) added to allopurinol in patients with serum uric acid (sUA) above target in a 12-month, randomised, phase III trial.
Methods Patients on allopurinol ≥300 mg (≥200 mg in moderate renal impairment) had sUA level of ≥6.5 mg/dL (≥387 µmol/L) at screening and two or more gout flares in the prior year. Primary end point was the proportion of patients achieving sUA level of <6.0 mg/dL (<357 µmol/L) (month 6). Key secondary end points were mean gout flare rate requiring treatment (months 7 through 12) and proportions of patients with complete resolution of one or more target tophi (month 12). Safety assessments included adverse events and laboratory data.
Results Patients (n=610) were predominantly male, with mean (±SD) age 51.2±10.90 years, gout duration 11.5±9.26 years and baseline sUA of 6.9±1.2 mg/dL (410±71 µmol/L). Lesinurad at 200 and 400 mg doses, added to allopurinol, significantly increased proportions of patients achieving sUA target versus allopurinol-alone therapy by month 6 (55.4%, 66.5% and 23.3%, respectively, p<0.0001 both lesinurad+allopurinol groups). In key secondary end points, there were no statistically significant treatment-group differences favouring lesinurad. Lesinurad was generally well tolerated; the 200 mg dose had a safety profile comparable with allopurinol-alone therapy. Renal-related adverse events occurred in 5.9% of lesinurad 200 mg+allopurinol, 15.0% of lesinurad 400 mg+allopurinol and 4.9% of allopurinol-alone groups, with serum creatinine elevation of ≥1.5× baseline in 5.9%, 15.0% and 3.4%, respectively. Serious treatment-emergent adverse events occurred in 4.4% of lesinurad 200 mg+allopurinol, in 9.5% of lesinurad 400 mg+allopurinol and in 3.9% of allopurinol-alone groups, respectively.
Conclusion Lesinurad added to allopurinol demonstrated superior sUA lowering versus allopurinol-alone therapy and lesinurad 200 mg was generally well tolerated in patients with gout warranting additional therapy.
Trial registration number NCT01493531.
- Gout
- Inflammation
- Outcomes research
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Gout is an inflammatory arthritis characterised by the deposition of monosodium urate (MSU) crystals in the joints, tendons and other connective tissues. Crystal deposition secondary to long-standing hyperuricemia can be reversed by lowering the concentration of serum uric acid (sUA) below the MSU saturation point—leading, in the long term, to the potential disappearance of signs and symptoms of gout. As a result, current management guidelines recommend maintenance of sUA to <6.0 mg/dL (<357 µmol/L) in patients with gout.1–3
Allopurinol is recommended as a first-line urate-lowering therapy (ULT).2 ,4 However, clinical trials have demonstrated that >50% of patients do not achieve sustained reductions in sUA at the most commonly used allopurinol dose of 300 mg.5–8 Lesinurad (RDEA594) is a novel, selective uric acid reabsorption inhibitor (SURI) for treatment of gout in combination with xanthine oxidase inhibitors. Lesinurad inhibits URAT1, a uric acid transporter responsible for the reabsorption of uric acid from the renal tubular lumen.9–11 Lesinurad in combination with allopurinol therefore provides a dual mechanism for sUA lowering—an increase in excretion of uric acid and a reduction in urate production.
Clinical studies have demonstrated that lesinurad in combination with allopurinol reduces mean sUA concentrations and increases proportions of patients who achieve sUA targets.12–14 The current phase III study—Combining Lesinurad with Allopurinol Standard of Care in Inadequate Responders (CLEAR 2)—is one of two replicate, randomised, double-blind, placebo-controlled, multicentre studies to investigate lesinurad in combination with allopurinol in patients with gout. CLEAR 1 was performed within the USA, included 603 patients with gout and provided outcomes similar to the CLEAR 2 study.15
Methods
Study design
CLEAR 2 was an international, phase III trial to investigate the efficacy and safety of two lesinurad doses (200 or 400 mg oral, once daily) in combination with allopurinol, versus allopurinol combined with placebo (the control arm), in patients demonstrating inadequate response to standard-of-care allopurinol (ClinicalTrials.gov Identifier: NCT01493531). The study was conducted in 12 countries in Europe, North America, South Africa, Australia and New Zealand between December 2011 and July 2014.
CLEAR 2 included a screening period of approximately 28 days, including a run-in of approximately 14 days on gout flare prophylaxis and 12-month double-blind treatment (figure 1). The study was conducted in accordance with Independent Ethics Committee E6 Good Clinical Practice, the Declaration of Helsinki (October 2008) and all applicable local regulatory requirements.

CLEAR 2 trial design is shown. *200 mg permitted for renally impaired. Maximum allopurinol dose: 800 or 900 mg, according to local label. Randomisation was stratified at day −7 by renal function (ie, estimated eCrCl ≥60 vs <60 mL/min, calculated by the Cockcroft-Gault formula using ideal body weight) and by tophus status during screening (ie, one or more tophus versus no tophi). eCrCl, estimated creatinine clearance; sUA, serum uric acid.
Patients
Male or female patients aged 18–85 years with a diagnosis of gout, body mass index <45 kg/m2, inadequate hypouricaemic response to standard-of-care allopurinol and two or more gout flares in the previous 12 months were eligible for study inclusion. Patients were included if they met the 1977 American Rheumatism Association preliminary classification criteria for gout.16 Patients were required to have received allopurinol as the sole ULT for ≥8 weeks prior to screening at a dose assessed medically appropriate by the treating physician (minimum 300 mg/day (200 mg in moderate renal impairment)17 up to 800 or 900 mg, depending on locally approved dose). sUA was required to be ≥6.5 mg/dL (≥387 µmol/L) at screening and ≥6.0 mg/dL (≥357 µmol/L) approximately 7 days prior to start of treatment on day 1.
Patients with estimated creatinine clearance (eCrCl) <30 mL/min were excluded from study. Patients with a history of kidney stones were permitted. Complete exclusion criteria are included in the online supplementary material 1.
supplementary data
Study medications
Eligible patients were randomised by double-blind method to one of three treatment groups (lesinurad 200 mg, lesinurad 400 mg or placebo) in 1:1:1 ratio, added to continued treatment with allopurinol at pre-study dose. Randomisation at study sites used a centralised Interactive Voice Response System/Interactive Web Response System.
Doses of lesinurad or matching placebo were taken once daily in the morning with food and one cup of water. Compliance was assessed from dispensing records and verification of returned medication packaging. Concomitant medication use was recorded at each study visit.
Gout flare prophylaxis was initiated at day −14, that is, the same time as sponsor-provided allopurinol. Prophylaxis consisted of colchicine (0.5 or 0.6 mg/day, as locally available) or a non-steroidal anti-inflammatory drug (NSAID, dosed according to local prescribing practice, with or without proton-pump inhibitor) for patients who were intolerant to or had contraindications to colchicine. Gout flare prophylaxis was continued through month 5, unless patients became intolerant or developed toxicity to prophylaxis.
Patients were encouraged to drink 2 L of fluid a day and remain well hydrated, following American College of Rheumatology guidelines for management of gout.2
Assessments
Efficacy assessments
The primary efficacy end point was the proportion of patients in each treatment group with sUA <6.0 mg/dL (<357 µmol/L) by month 6. Other sUA-related end points included proportions of patients with sUA <6.0 mg/dL (<357 µmol/L), <5.0 mg/dL (<297 µmol/L) and <4.0 mg/dL (<238 µmol/L) and mean absolute and mean percentage changes from baseline in sUA at each visit.
Two key secondary end points included: (1) mean rate of gout flares requiring treatment for the 6-month period from end of month 6 to end of month 12, reported on a daily electronic patient diary. This key secondary end point included only clinically relevant gout flares, which were those requiring either an increase in current medication or new medication and (2) proportion of patients with target tophi at baseline who experienced complete resolution of one or more target tophi by month 12, that is, 100% decrease in tophus area. Target tophi (up to five per patient) were tophi on the hands/wrists and/or feet/ankles measured by digital Vernier callipers at ≥5 and ≤20 mm in longest diameter.18 Permitted treatments for gout flares were colchicine, analgesics and/or anti-inflammatory medications, including oral and intra-articular corticosteroids.
Safety assessments
Safety assessments included treatment-emergent adverse events (TEAEs; coded by Medical Dictionary for Regulatory Activities (V.14.0)), clinical laboratory data, physical examination, ECG and vital signs. Adverse events (AEs) of special interest included renal and cardiovascular (CV) safety assessments.
Assessments of renal safety included renal-related and kidney stone TEAEs (see online supplementary material 2) and clinical laboratory data, including serum creatinine (sCr), creatine kinase, urine protein-to-creatinine ratio and eCrCl levels. CV safety was of special interest because of the known high rates of CV risk factors in patients with gout.19 ,20 An independent Cardiovascular Events Adjudication Committee (CEAC) routinely assessed AEs for potential CV relationship, with categorisation into major adverse CV events (MACEs) and non-MACE end points (see online supplementary material 3).21
Statistical analyses
Comparisons of response proportions based on sUA level between each lesinurad plus allopurinol group and the allopurinol-alone group were performed using the Cochran-Mantel-Haenszel (CMH) test statistic, stratified by day −7 renal function and tophus status during screening. A Bonferroni correction was used for the primary end point for each of the two treatment comparisons with allopurinol-alone therapy at an α level of 0.025. Testing of the key secondary end points hierarchically at an α level of 0.05 was gated on both dose contrasts being statistically significant for the primary end point. If only one of the primary end point dose contrasts was significant, then α=0.025 for each key secondary end point within the surviving dose. All other efficacy end points were evaluated at α=0.05 (nominal p value), two-sided, without multiplicity adjustment. Results for the primary end point of sUA response are expressed as proportions and p values. Patients with missing values at month 6 or month 12 for any reason were considered non-responders (non-responder imputation, NRI).22 Key secondary end points were analysed using negative binomial regression (gout flares) or CMH test (tophus response). Mean rates of gout flares were adjusted for day −7 renal function, tophus status at screening and length of exposure to randomised study medication. The time points and analytical methods used in the study were agreed with multiple regulatory agencies.
Safety data are listed by treatment arm and are not subjected to statistical testing. TEAEs are coded by system organ class and preferred term and are listed according to incidence, severity, relation to study medication and relation to discontinuation. To better identify potential clinically relevant changes in sCr related to lesinurad by minimising discrepancies due to intra-subject variability, baseline sCr was defined as the highest value within 14 days prior to first dose of study medication. Relative increase in sCr (ie, ≥1.5× and ≥2.0× the baseline level at any time) was selected as the most clinically relevant sCr assessment.23 ,24 Resolution of sCr elevation was defined as an sCr value returned to ≤1.2× baseline.
A sample size of approximately 600 patients was planned to be recruited, for an allocation of approximately 200 patients to each treatment arm. This sample size was calculated to provide greater than 90% power to detect a difference in response rate between treatment groups if the allopurinol-alone group had a 30% response rate and the lesinurad groups had response rates as low as 48% using Fisher's exact test, adjusting for multiplicity with α=0.025, two-sided, for each test.
All randomised patients who received at least one dose of study medication were included in the intent-to-treat (ITT) population, which was the primary population for efficacy and safety assessments.
Results
Patient disposition
Of the 2199 patients screened, 611 were randomised at 152 sites. Of the 611 randomised patients, 610 received at least one dose of study medication (figure 2).

Patient disposition is shown. aScreened was defined as signing an informed consent form; b2 deaths reported for non-randomised patients during screening and ccompleted the study with or without completing randomised study medication. One additional death occurred in the LESU 400 mg+ALLO group. The subject experienced a serious adverse event and withdrew from the study. The primary reason for study withdrawal was reported as ‘adverse event’. Of the 1538 screen failures, 1183 were related to inclusion criteria, 252 to exclusion criteria, 94 to both inclusion and exclusion criteria and 9 to other.
ALLO, allopurinol; LESU, lesinurad.
Demographic characteristics and clinical history
Demographics and baseline disease characteristics were similar between treatment groups (table 1). Patients generally had long-standing symptomatic gout (mean (±SD) time since diagnosis 11.5±9.3 years) and elevated baseline sUA (mean 6.9±1.2 mg/dL (410±71 µmol/L)), with high rates of one or more predefined comorbidities (ie, CV risk factors or kidney stones) at 79.2%.
Demographic and baseline disease characteristics (intent-to-treat population)
Most patients (84.1%) received allopurinol at a daily dose of 300 mg, with 6.6% receiving <300 mg and 9.3% receiving >300 mg; the overall dose range was 200–900 mg.
Study medications
Proportions of patients exhibiting ≥80% compliance with study medication were 97.6%, 94.1% and 94.5% in the allopurinol-alone, lesinurad 200 mg+allopurinol and lesinurad 400 mg+allopurinol groups, respectively.
Efficacy assessments
Primary end point of sUA response and secondary sUA end points
Proportions of patients achieving sUA level of <6.0 mg/dL (<357 µmol/L) by month 6 (the primary end point) were 23.3%, 55.4% and 66.5% in the allopurinol-alone, lesinurad 200 mg+allopurinol and lesinurad 400 mg+allopurinol groups, respectively, using NRI—significant differences were identified for both lesinurad+allopurinol groups versus allopurinol-alone (p<0.0001; CMH test) (figure 3).

Proportions of patients achieving sUA target of <6.0 mg/dL (<357 µmol/L), <5.0 mg/dL (<297 µmol/L) and <4.0 mg/dL (<238 µmol/L), by months 6 and 12 (ITT population) are shown. Primary end point: proportion of patients achieving sUA target of <6.0 mg/dL (<357 µmol/L) by month 6. *p<0.0001. Note: Subjects missing sUA results were treated as non-responders. All comparisons used a two-sided Cochran-Mantel-Haenszel test stratified by day −7 renal function and tophus status during screening (randomised stratification factor values), with non-responder imputation and adjustment for multiple comparisons for the primary end point (Bonferroni correction). ALLO, allopurinol; ITT, intention to treat; LESU, lesinurad; sUA, serum uric acid.
Subgroup analyses based on age, sex, race, baseline sUA, comorbidities, renal function and thiazide diuretic use provided results consistent with primary analysis of the ITT population (see online supplementary material 4 for renal function and diuretic analyses).
Proportions of patients achieving the sUA target of <6.0 mg/dL (<357 µmol/L) were greater in the lesinurad 200 mg+allopurinol and lesinurad 400 mg+allopurinol versus allopurinol-alone group at all monthly assessments from month 1 to month 12 (nominal p<0.0001, all comparisons). Proportions of patients achieving sUA level of <5.0 mg/dL (<297 µmol/L) and <4.0 mg/dL (<238 µmol/L) were also greater in both lesinurad+allopurinol groups versus allopurinol-alone group at each monthly visit (sUA <5.0 mg/dL (<297 µmol/L): nominal p<0.0001, both comparisons; sUA <4.0 mg/dL (<238 µmol/L): nominal p<0.0001, both comparisons, except p<0.01 at month 1, lesinurad 200 mg+allopurinol). Figure 3 shows proportions of patients at each sUA threshold by months 6 and 12.
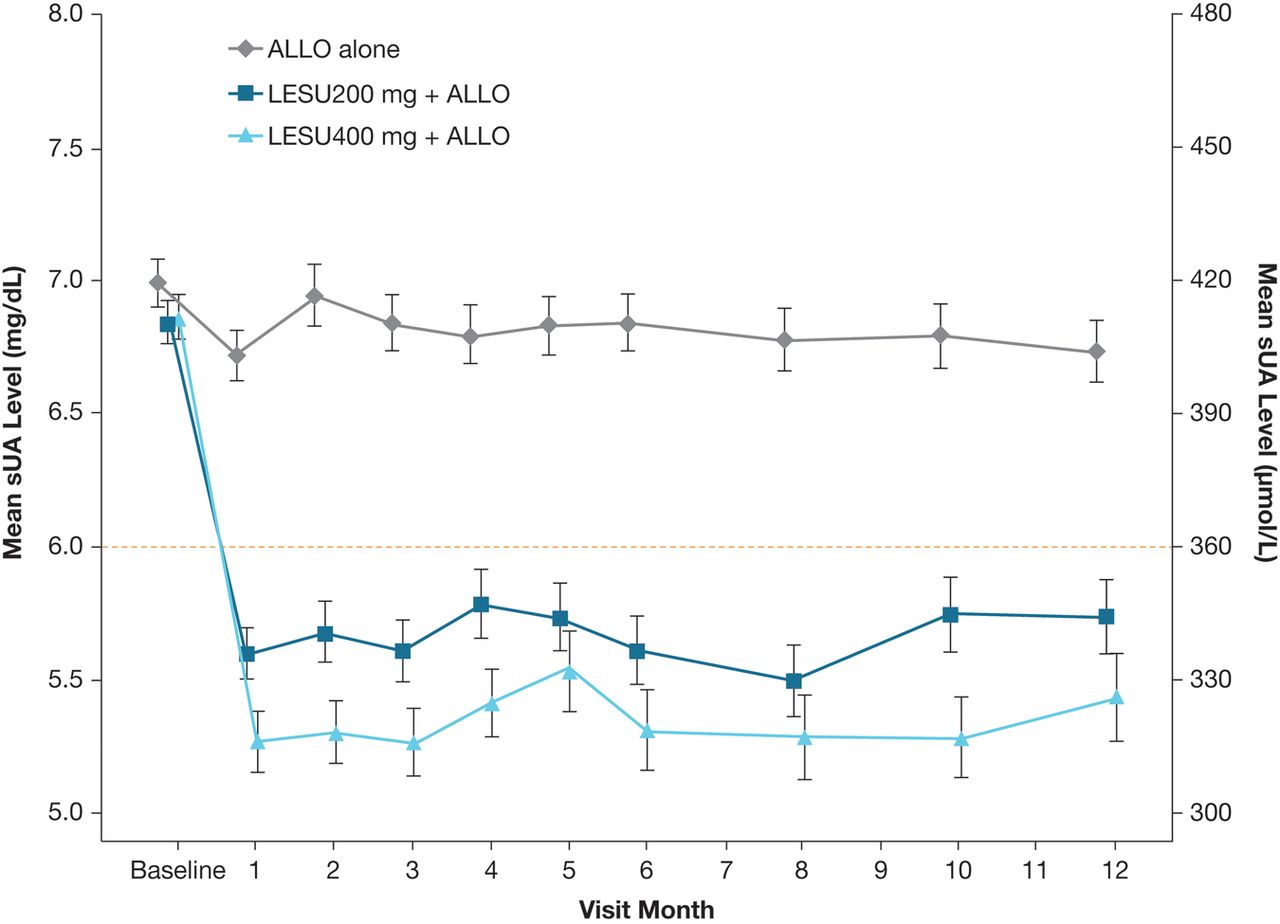
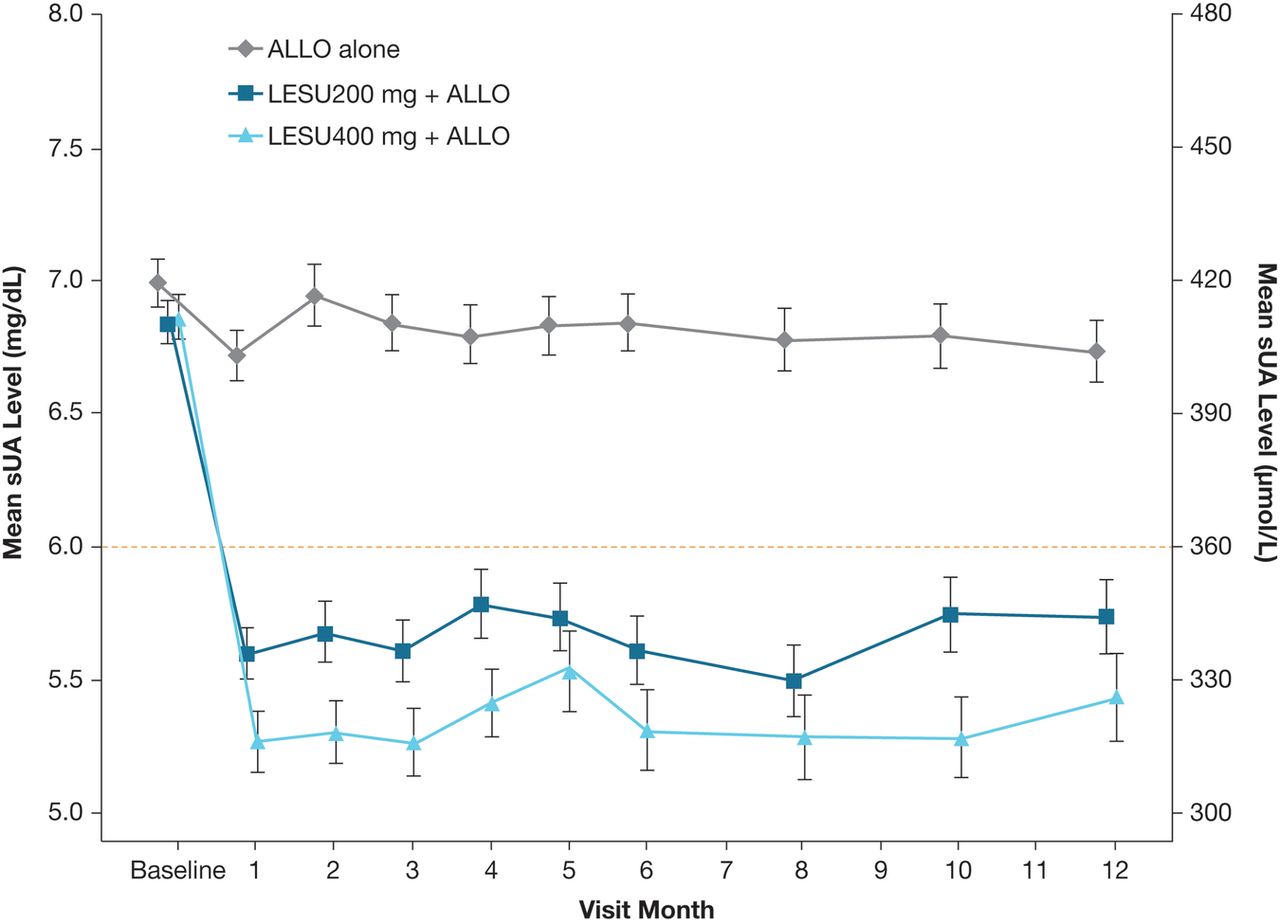
Mean sUA levels were lower in both lesinurad+allopurinol groups versus allopurinol-alone group at all time points (nominal p<0.001, both comparisons compared with allopurinol-alone therapy) (figure 4).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Graph showing the mean (SE) sUA levels by visit (observed cases, intent-to-treat population). Mean change from baseline for each active treatment group was compared with the ALLO-alone group using analysis of covariance, with p<0.001 at each time point. ALLO, allopurinol; LESU, lesinurad; sUA, serum uric acid.
Secondary end point: gout flares requiring treatment
The gout flare rate and the proportions of patients with gout flares requiring treatment were low and similar in all groups throughout the study. Mean (±SE) rates of gout flares requiring treatment from the end of month 6 to end of month 12 were 0.83±0.13 for allopurinol-alone group versus 0.73±0.12 and 0.77±0.13, respectively, in the lesinurad 200 mg+allopurinol and lesinurad 400 mg+allopurinol groups (p=0.57 and 0.75 vs allopurinol-alone group). Proportions of patients with a gout flare requiring treatment through the study are shown in online supplementary figure S2.
Secondary end point: tophus resolution
The numbers of patients with one or more target tophi at baseline were low: 33, 35 and 29 in the allopurinol-alone, lesinurad 200 mg+allopurinol and lesinurad 400 mg+allopurinol groups, respectively. In these respective groups, 33.3%, 31.4% and 27.6% of patients achieved complete resolution of one or more target tophi by month 12 (p>0.05, both lesinurad+allopurinol groups vs allopurinol-alone group).
Safety assessments
Adverse events
TEAEs were reported in 70.9%, 74.5% and 80.5% of the allopurinol-alone, lesinurad 200 mg+allopurinol and lesinurad 400 mg+allopurinol groups, respectively (table 2). The majority of TEAEs in each group had a maximum severity of grade 1 or grade 2, based on Rheumatology Common Toxicity Criteria.25 The most common individual TEAEs—reported for allopurinol-alone, lesinurad 200 mg+allopurinol and lesinurad 400 mg+allopurinol groups, respectively—were upper respiratory tract infection (10.2%, 6.9%, 15.0%), hypertension (4.9%, 8.3%, 8.0%), arthralgia (4.4%, 11.8%, 3.0%), increased blood creatinine (3.4%, 3.9%, 9.5%) and diarrhoea (3.4%, 4.9%, 7.0%). The most common grade 3 or grade 4 TEAEs in these respective groups were increased blood creatine kinase (1.5%, 0.5%, 1.5%) and myocardial infarction (MI) (0%, 0%, 1.5%).
Overall summary of TEAEs (safety population)
Serious TEAEs were reported in 3.9%, 4.4% and 9.5% of patients, respectively (table 2). Two deaths occurred in the lesinurad 400 mg+allopurinol group (pulmonary oedema and gastric cancer, respectively). TEAEs led to study-medication discontinuation in 5.3%, 3.4% and 9.5% of the allopurinol-alone, lesinurad 200 mg+allopurinol and lesinurad 400 mg+allopurinol groups, respectively; the most common TEAE leading to discontinuation was increased blood creatinine (1.0%, 0% and 2.5%, respectively).
Renal safety analyses
Renal-related TEAEs occurred in 4.9%, 5.9% and 15.0%, of allopurinol-alone, lesinurad 200 mg+allopurinol and lesinurad 400 mg+allopurinol groups, respectively. The most common renal-related TEAEs in these respective groups were increased blood creatinine (3.4%, 3.9%, 9.5%), increased blood urea (0%, 2.0%, 1.5%) and renal failure (0.5%, 1.0%, 1.5%). One patient (0.5%) in the allopurinol-alone group experienced a serious renal-related TEAE, versus no patients in the lesinurad 200 mg+allopurinol and two patients (1.0%) in the lesinurad 400 mg+allopurinol group. Kidney stone TEAEs were reported in 0.5%, 0% and 3.0%, respectively.
sCr elevation ≥1.5× baseline occurred in 3.4% (n=7), 5.9% (n=12) and 15.0% (n=30) of allopurinol-alone, lesinurad 200 mg+allopurinol and lesinurad 400 mg+allopurinol groups, respectively. sCr elevation ≥1.5× was transient and reversible in most cases and the majority of sCr elevations resolved by the time of the next assessment; there were three unresolved sCr elevations in the allopurinol-alone group at last visit versus none in the lesinurad 200 mg+allopurinol and seven in the lesinurad 400 mg+allopurinol group (see online supplementary table S1). sCr elevation ≥2.0× baseline occurred in 0%, 2.0% (n=4) and 8.0% (n=16) of patients, respectively. Again, most elevations ≥2.0× baseline were transient and reversible; no sCr elevations ≥2.0× were unresolved at last visit in the lesinurad 200 mg+allopurinol group and five cases were unresolved in the lesinurad 400 mg+allopurinol group. In approximately two-thirds of sCr elevations, resolution occurred while patients continued on study medication.
In all treatment groups, proportions of patients with an sCr elevation ≥1.5× baseline tended to be higher for patients (1) who were taking an NSAID than colchicine; (2) who did not achieve target sUA at month 6 versus responders and (3) who had one or more tophi at screening versus those without tophi, although small subgroup sizes render interpretation difficult (see online supplementary table S2). There was no apparent association between sCr elevation and baseline renal function or other concomitant medications.
Renal function remained stable across the treatment groups, as measured by mean (SD) changes in eCrCl, from baseline to last value. Mean (±SD) changes in eCrCl in the allopurinol-alone, lesinurad 200 mg+allopurinol and lesinurad 400 mg+allopurinol groups were 3.0±9.7, −0.5±11.5 and −5.7±13.9 mg/dL, respectively, from baseline to last value on treatment and were 1.8±11.7, 2.7±10.0 and 1.1±24.2 mg/dL from baseline to last value off treatment at follow-up (in patients not entering a separate extension study, n=133).
CV safety analyses
TEAEs were adjudicated as CV events in 5.3% (n=11 patients), 3.9% (n=8 patients) and 3.0% (n=6 patients) of allopurinol-alone, lesinurad 200 mg+allopurinol and lesinurad 400 mg+allopurinol groups, respectively. CEAC-adjudicated criteria for MACE were met by three patients (four events, including three MIs and one death due to pulmonary oedema), all in the lesinurad 400 mg+allopurinol group. Non-MACE CV end points were reported in five patients (five events), two patients (two events) and no patients, respectively.
Other clinical laboratory tests and vital signs
Clinical laboratory results (excluding renal laboratory results, reported above) and urinalysis were comparable between treatment groups. Elevations in creatine kinase >5× upper limit of normal were in 5.3%, 2.0% and 3.0% of allopurinol-alone, lesinurad 200 mg+allopurinol and lesinurad 400 mg+allopurinol groups, respectively. There were no notable changes in vital signs.
Discussion
Allopurinol at the 300 mg dose is frequently unable to achieve target sUA levels.5–8 Guidelines recommend increasing the allopurinol dose above 300 mg/day to attain target sUA, but this happens rarely in practice, in part due to physician's concerns over safety of doses >300 mg.4 ,6 ,25–29 Other management options include switching from allopurinol to febuxostat, or adding a uricosuric to allopurinol, based on evidence from earlier, small trials.30–32 CLEAR 2 and the similarly designed CLEAR 115 were the initial large studies to validate a combination approach using a URAT1 inhibitor that inhibits uric acid reabsorption (ie, lesinurad) with allopurinol.
In CLEAR 2, lesinurad at both doses (200 or 400 mg) combined with continued allopurinol significantly increased the proportions of patients achieving sUA target of <6.0 mg/dL (<357 µmol/L) by month 6 (p<0.0001), with more than twice as many patients reaching goal versus allopurinol-alone therapy. Onset of sUA reduction in the lesinurad groups was rapid, with significant differences from allopurinol-alone group by first assessment at month 1. The significant increase in proportions of patients who achieved sUA target in both lesinurad+allopurinol groups versus allopurinol-alone group was sustained over the 12-month study. Consistent response rates were observed, irrespective of renal function or thiazide diuretic use.
There were no statistically significant differences favouring lesinurad treatment in the rates of gout flare requiring treatment or complete resolution of tophi, which occurred at low incidences at baseline and during study. In relation to these key secondary end points, treatment may be required for more than 12 months for the full effects to be observed.33
Lesinurad was generally well tolerated, particularly at the 200 mg dose, where the TEAE and serious TEAE profiles were comparable with the allopurinol-alone group. Higher TEAE incidences were seen in the lesinurad 400 mg+allopurinol group. Renal-related TEAEs occurred at similar incidences in the lesinurad 200 mg+allopurinol and allopurinol-alone groups, with a higher incidence in the lesinurad 400 mg+allopurinol than lesinurad 200 mg+allopurinol group; the lesinurad 400 mg+allopurinol group also showed a higher incidence of sCr elevation. The majority of sCr elevations resolved by the next assessment and in most cases without interruption in study medication. Mean renal function did not differ between the treatment groups both before and after treatment. The mechanism of sCr elevation associated with lesinurad may be via increased excretion of urinary uric acid, which has the potential to induce uric acid microcrystallisation in the renal tubules. Urine protein-to-creatinine ratio and urinalyses did not change during the study, suggesting that sCr elevation was not associated with renal parenchymal sequelae. Patients with unresolved sCr elevations showed no defining characteristics compared with those whose sCr elevation resolved.
Other therapies which inhibit URAT1 have been associated with development of kidney stones.34 ,35 The lack of increase in kidney stone numbers during lesinurad therapy is potentially because of concomitant allopurinol use, which reduces uric acid production.36 ,37 The rate of nephrolithiasis may also have been influenced by timing of lesinurad administration, as once-daily dosing in the morning increases urinary uric acid at a time when urine volume and urine pH are highest and the potential for uric acid precipitation is lowest.38 ,39
Prescribing information for the approved dose of lesinurad 200 mg recommends assessment of renal function prior to initiation of therapy and periodically thereafter, particularly in patients whose CrCl is 30–<45 mL/min, with discontinuation recommended if CrCl is persistently <30 mL/min (ie, severe renal impairment). Lesinurad is also contraindicated in subjects with end-stage renal disease, kidney transplant recipients or patients on dialysis.
CV comorbidities and risk factors were present in approximately 80% of patients, reflecting the high rates of CV disease in patients with gout.40–42 The proportions of patients with TEAEs classified as CV events during study were low and similar in treatment groups. Incidences of MACE events—that is, serious CV events including CV deaths, non-fatal MI and non-fatal stroke—were similarly low. Three patients experienced MACE in the study, all receiving lesinurad 400 mg. Low rates of MACE events during gout treatment were also reported in the open-label Long-term Allopurinol Safety Study Evaluating Outcomes in Gout Patients (LASSO) study, which reported a rate of 0.58% over 6 months for MACE during allopurinol treatment (incidence rate 1.42/100 patient-years).6
Limitations of CLEAR 2 include the limited data on allopurinol doses >300 mg, the relatively low proportion of women enrolled, low number of patients with evaluable tophi and the relatively short-term follow-up period that limits the ability to adequately study flares and tophi. Rates of gout flares and tophus resolution over the longer term are being investigated in an extension study (NCT01808131).
In conclusion, lesinurad (200 and 400 mg), a novel SURI, in combination with allopurinol significantly increased the proportion of patients achieving the target sUA of <6.0 mg/dL (<357 µmol/L) by month 6 and other sUA end points compared with allopurinol-alone therapy. There were no statistically significant treatment-group differences favouring lesinurad for rate of gout flares or complete tophus resolution. The combination therapy was generally well tolerated, particularly at the 200 mg lesinurad dose approved by the US Food and Drug Administration and European Medicines Agency, except for higher incidences of predominantly reversible sCr elevation compared with allopurinol-alone therapy. There were no cases of unresolved sCr elevation ≥1.5× in the lesinurad 200 mg+allopurinol group, versus three unresolved cases in the allopurinol-alone group and seven in the lesinurad 400 mg+allopurinol group. By using a dual mechanism approach to reduce sUA, combination therapy with lesinurad and allopurinol represents a treatment option for patients with gout inadequately controlled on allopurinol-alone therapy.
Acknowledgments
Editorial support for this manuscript was provided by Bill Wolvey of PAREXEL, which was funded by AstraZeneca.
References
Supplementary materials
Lay summary
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Tore K Kvien
JK and CS are former employees.
Contributors TB, RTK, PPK and AS: Criterion 1: (1) substantial contributions to study conception and design and/or (2) substantial contributions to analysis and interpretation of data; criterion 2: drafting the article or revising it critically for important intellectual content and criterion 3: final approval of the version of the article to be published. JK, MF, NB, CS and SB: Criterion 1: (1) substantial contributions to study conception and design and/or (2) substantial contributions to acquisition of data and/or (3) substantial contributions to analysis and interpretation of data; criterion 2: drafting the article or revising it critically for important intellectual content and criterion 3: final approval of the version of the article to be published. SA: criterion 1: (1) substantial contributions to acquisition of data and/or (2) substantial contributions to analysis and interpretation of data; criterion 2: drafting the article or revising it critically for important intellectual content and criterion 3: final approval of the version of the article to be published.
Funding This clinical study was funded by Ardea Biosciences, a member of the AstraZeneca group. The study sponsor had a role in the design and conduct of the study; collection, management, analysis and interpretation of the data and review and approval of the manuscript.
Competing interests TB: grant/research support from Ipsen, Menarini and consultant for AstraZeneca, Ipsen, Menarini, Novartis, Savient, Sobi, Takeda and Cymabay. RTK: consultant for AstraZeneca, Crealta Pharmaceuticals and Takeda. PPK: research grant: AstraZeneca. JK (former employee), MF, NB, CS (former employee) and SB: full-time employees of Ardea Biosciences, a member of the AstraZeneca Group. SA: full-time employee of AstraZeneca Pharmaceuticals. AS: consultant for Novartis, AstraZeneca, Menarini.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.