Article Text

Abstract
Objectives Tofacitinib is an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis (RA). To further assess the potential role of Janus kinase inhibition in the development of malignancies, we performed an integrated analysis of data from the tofacitinib RA clinical development programme.
Methods Malignancy data (up to 10 April 2013) were pooled from six phase II, six Phase III and two long-term extension (LTE) studies involving tofacitinib. In the phase II and III studies, patients with moderate-to-severe RA were randomised to various tofacitinib doses as monotherapy or with background non-biological disease-modifying antirheumatic drugs (DMARDs), mainly methotrexate. The LTE studies (tofacitinib 5 or 10 mg twice daily) enrolled patients from qualifying prior phase I, II and III index studies.
Results Of 5671 tofacitinib-treated patients, 107 developed malignancies (excluding non-melanoma skin cancer (NMSC)). The most common malignancy was lung cancer (n=24) followed by breast cancer (n=19), lymphoma (n=10) and gastric cancer (n=6). The rate of malignancies by 6-month intervals of tofacitinib exposure indicates rates remained stable over time. Standardised incidence ratios (comparison with Surveillance, Epidemiology and End Results) for all malignancies (excluding NMSC) and selected malignancies (lung, breast, lymphoma, NMSC) were within the expected range of patients with moderate-to-severe RA.
Conclusions The overall rates and types of malignancies observed in the tofacitinib clinical programme remained stable over time with increasing tofacitinib exposure.
- Rheumatoid Arthritis
- Treatment
- DMARDs (synthetic)
- Inflammation
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Video abstract
Introduction
Chronic inflammation and autoimmune diseases are associated with the development of malignancies.1 ,2 In patients with rheumatoid arthritis (RA), regardless of treatment regimens, some malignancies such as Hodgkin's and non-Hodgkin's lymphoma, leukaemia, myeloma and lung cancer occur more frequently than in the general population.1 ,3 The relationship between malignancies and RA is complex as the immune response and some RA treatments (such as non-steroidal anti-inflammatory drugs and glucocorticoids) can also affect malignancy rates.1 4 Furthermore, in addition to the malignancy risk associated with RA, there is also a malignancy risk associated with treatments for chronic inflammation and autoimmune diseases that involve modulation of the immune system.5 ,6
Tofacitinib is an oral Janus kinase inhibitor for the treatment of RA. The efficacy and safety of tofacitinib 5 and 10 mg twice daily has been demonstrated in a variety of patient populations with moderate-to-severe active RA in phase II7–10 and phase III11–16 randomised controlled trials, and two long-term extension (LTE) studies.17 ,18
In the development of immunomodulatory agents with new mechanisms of action such as tofacitinib, there is a particular need for close monitoring of safety events of special interest, including malignancies, to uncover potential adverse drug reactions. Here we report pooled malignancy data from the tofacitinib RA clinical development programme.
Methods
Patients
Eligible patients aged ≥18 years with active, moderate-to-severe RA were enrolled globally from North America, Europe, Latin America and Asia (see online supplementary appendix for list of countries). Patients were required to have had an inadequate response to methotrexate (MTX) (NCT00413660;10 NCT00603512;19 ORAL Scan, NCT00847613;16 ORAL Standard, NCT0085338515), non-biological or biological disease-modifying antirheumatic drugs (DMARDs) (NCT00147498;7 NCT00550446;8 NCT00687193;9 ORAL Solo, NCT00814307;14 ORAL Sync, NCT0085654413) or tumour necrosis factor inhibitors (TNFi) (ORAL Step, NCT0096044011). One phase III study (ORAL Start, NCT0103968812) included MTX-naive patients and one phase II study (study NCT0105986420) had no criteria for prior DMARD exposure.
Exclusion criteria were similar across studies; patients with any history of or existing malignancy (other than adequately treated or excised non-metastatic basal cell or squamous cell cancer of the skin or cervical carcinoma in situ) were excluded. Patients who developed a malignancy (excepting adequately treated or excised non-metastatic basal cell or squamous cell cancer of the skin or cervical carcinoma in situ) were permanently discontinued from the study, but were followed up. Patients who developed non-melanoma skin cancer (NMSC) could remain in the study provided the NMSC was adequately treated or excised non-metastatic basal cell or squamous cell cancer of the skin or adequately treated cervical carcinoma in situ. Inclusion and exclusion criteria have been reported previously.7–20
Study design
Patients from six phase II7–10 ,19 ,20 and six phase III11–16 index studies, and two LTE studies,17 ,18 were included in the pooled phase II, III and LTE data. The LTE-only analysis comprised patients from two phase I,21 ,22 nine phase II7–10 ,19 ,20 ,23–25 and six phase III11–16 studies (see the online supplementary appendix for details on all index studies). The analysis reported here includes all patients with RA exposed to tofacitinib in the clinical development programme. As of April 2013, LTE17 ,18 and ORAL Start (NCT0103968812) data collection and analyses were ongoing, and study databases had not yet been locked. Further details on study design and index studies are given in the online supplementary appendix.
Outcome assessment and adjudication
Malignancies were identified and classified by review of investigator-reported adverse events (AEs), serious AEs and from the central laboratory histology review. A malignancy over-read process involved a centralised, external, blinded review of each biopsy case by ≥2 independent, board-certified pathologists. Discordance in opinion between local and central pathologists was uncommon and resolved by clinical review of all available data; results from both local and central pathologists were reported. Patients who had no biopsy slides available to central reading (25.8%; 335/1299) were reported according to the local pathology report. Patients with no pathology report (1.8%; 23/1299) were classified by malignancy type reported by the study investigator.
Statistical analysis
Malignancies in the tofacitinib clinical programme up to April 2013 were evaluated. For LTE analysis, patients were classified as background DMARDs or monotherapy based on the index study. Average total daily dose (TDD) of tofacitinib was calculated by adding all doses received by each patient, and dividing by the number of days a dose was received. The TDD average was used to assign LTE dose: TDD of <15 mg/day was considered 5 mg twice daily, TDD of ≥15 mg/day was considered 10 mg twice daily.
Incidence rates (IR) per 100 patient-years (py) of observation for all malignancies (excluding NMSC), lung cancer, breast cancer, lymphoma and NMSC were calculated by exposure and dose. Lung cancer, breast cancer and NMSC were selected as they are the most commonly occurring types; lymphoma was selected due to special interest in associated safety events in RA. IRs were based on number of patients with an event; for those with multiple malignancies, each patient was counted once for all malignancies, and separately for each event. The 95% CIs are based on maximum likelihood estimation, and on Exact Poisson adjusted for exposure time when IR is zero.
Standardised incidence ratios (SIRs) were calculated as the ratio of observed cancers to those expected based on background rates within general population samples; 95% CIs for SIRs were calculated following a Poisson distribution. Expected numbers of cancers for the calculations were based on age-specific and sex-specific rates from the US National Cancer Institute Surveillance and Epidemiology and End Results (SEER) database, 1992–2011.26 The SEER database does not include NMSC or carcinoma in situ of the cervix; therefore, these data were excluded from the tofacitinib data set for the calculation of SIRs only.
Results
Patients
Overall, 5671 patients in tofacitinib treatment groups were included in the analyses; 4204 patients received tofacitinib (across all doses) for >1 year, 3084 >2 years and 1948 >3 years. Total tofacitinib exposure was 12 664 py; overall median exposure was 2.35 years. Drug exposure data for tofacitinib, placebo and adalimumab are presented in figure 1A; baseline demography data are presented in table 1. In phase II and phase III studies, respectively, 657 (47.2%) and 1361 (35.9%) patients received tofacitinib as monotherapy and 736 (52.8%) and 2435 (64.1%) in combination with non-biological DMARDs.
Baseline demography data by treatment group for patients enrolled in the tofacitinib clinical programme (phase II and III, and LTE studies)
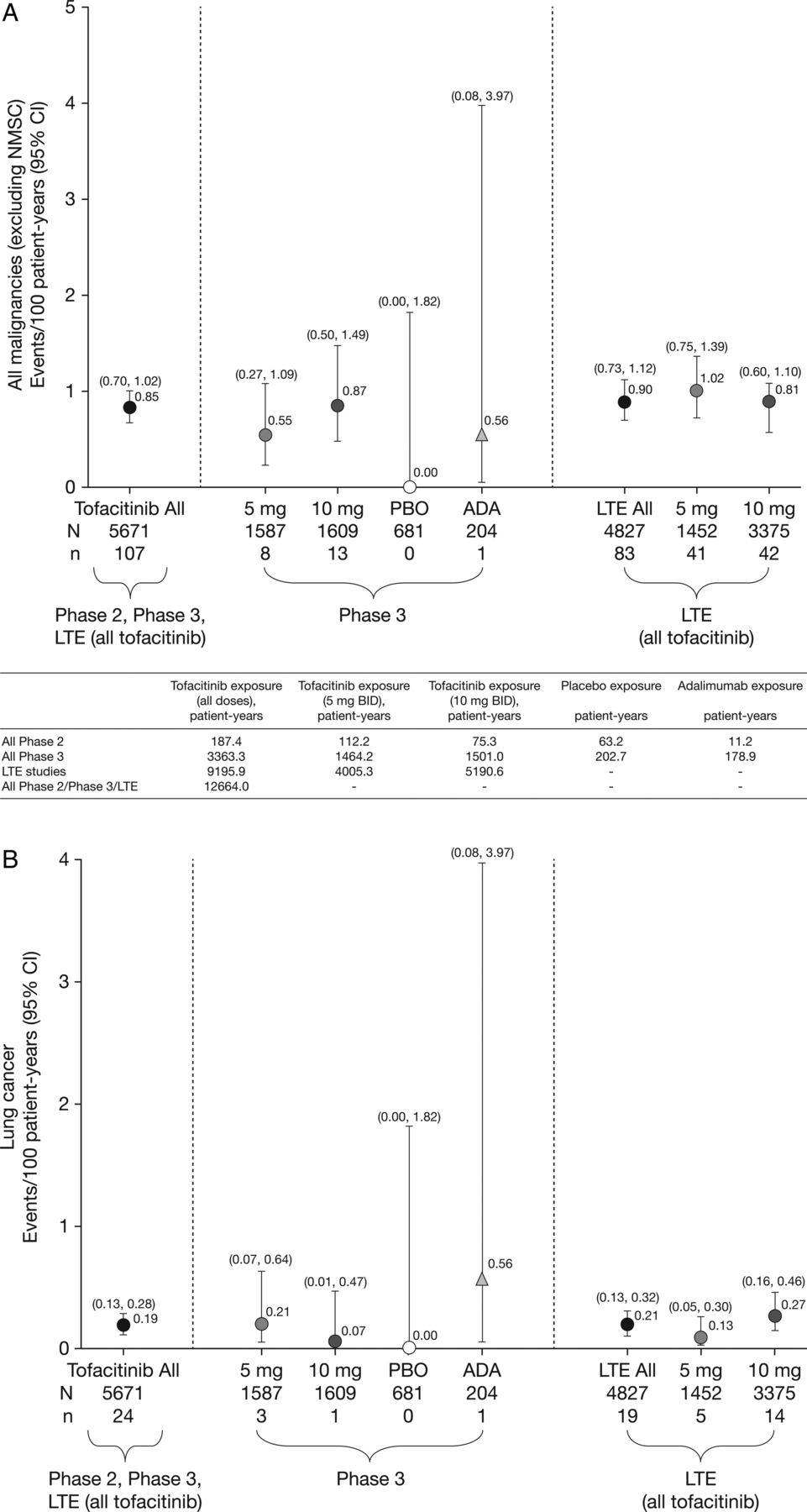


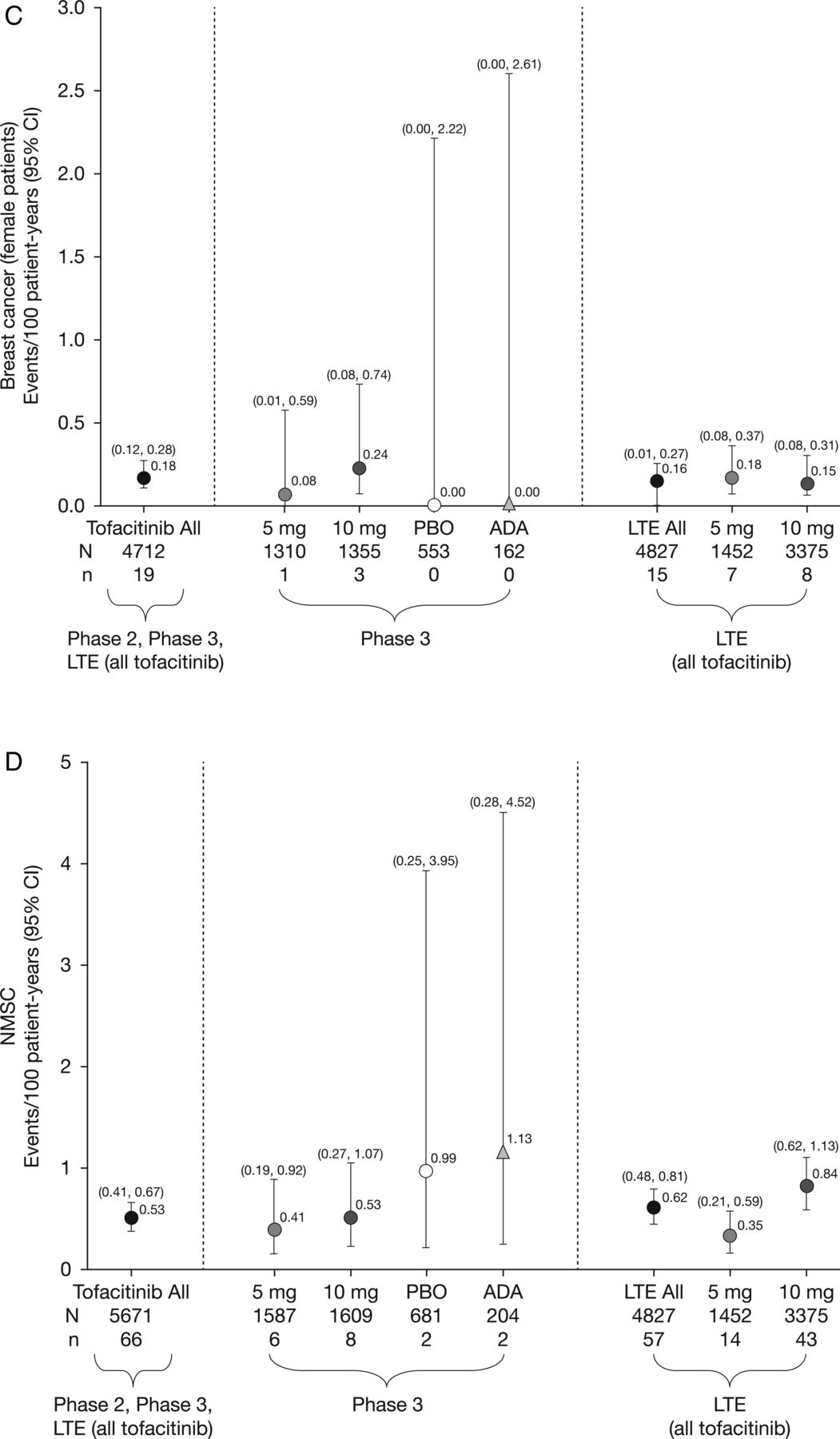
Incidence rates (and 95% CIs) for (A) all malignancies (excluding non-melanoma skin cancer (NMSC)), including drug exposure data; (B) lung cancer; (C) breast cancer (female patients); (D) NMSC and (E) lymphoma for all active treatments including dose groups. ADA, adalimumab; LTE, long-term extension; N, total number of patients in that group exposed to the study treatment; n, number of unique patients with malignancy event in each treatment group; PBO, placebo.
Observed malignancies
There were 107 tofacitinib-treated patients with malignancies (excluding NMSC); the most common was lung cancer (n=24) followed by breast cancer (n=19), lymphoma (n=10; for completeness, two additional cases are included—see ‘Lymphoma' section for details) and gastric cancer (n=6). Online supplementary appendix figure S2 lists each malignancy (excluding NMSC) in tofacitinib-treated patients observed per 6-month interval. In the adalimumab treatment group, there was one case of renal cell carcinoma (NCT00550446)8 in phase II and one of non-small-cell lung cancer (NSCLC; NCT00853385)15 in phase III. No malignancies (excluding NMSC) occurred in the phase II or III placebo groups (≤6-month duration). Eight patients receiving tofacitinib 5 mg twice daily, 13 receiving tofacitinib 10 mg twice daily and one each receiving adalimumab and MTX had more than one malignancy; the online supplementary appendix provides details on these and on patients with potential malignancies. IRs for all malignancies and selected malignancies for all active treatments, dose groups and placebo are shown in figure 1.
Overall rates of malignancies were stable over time (figure 2).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
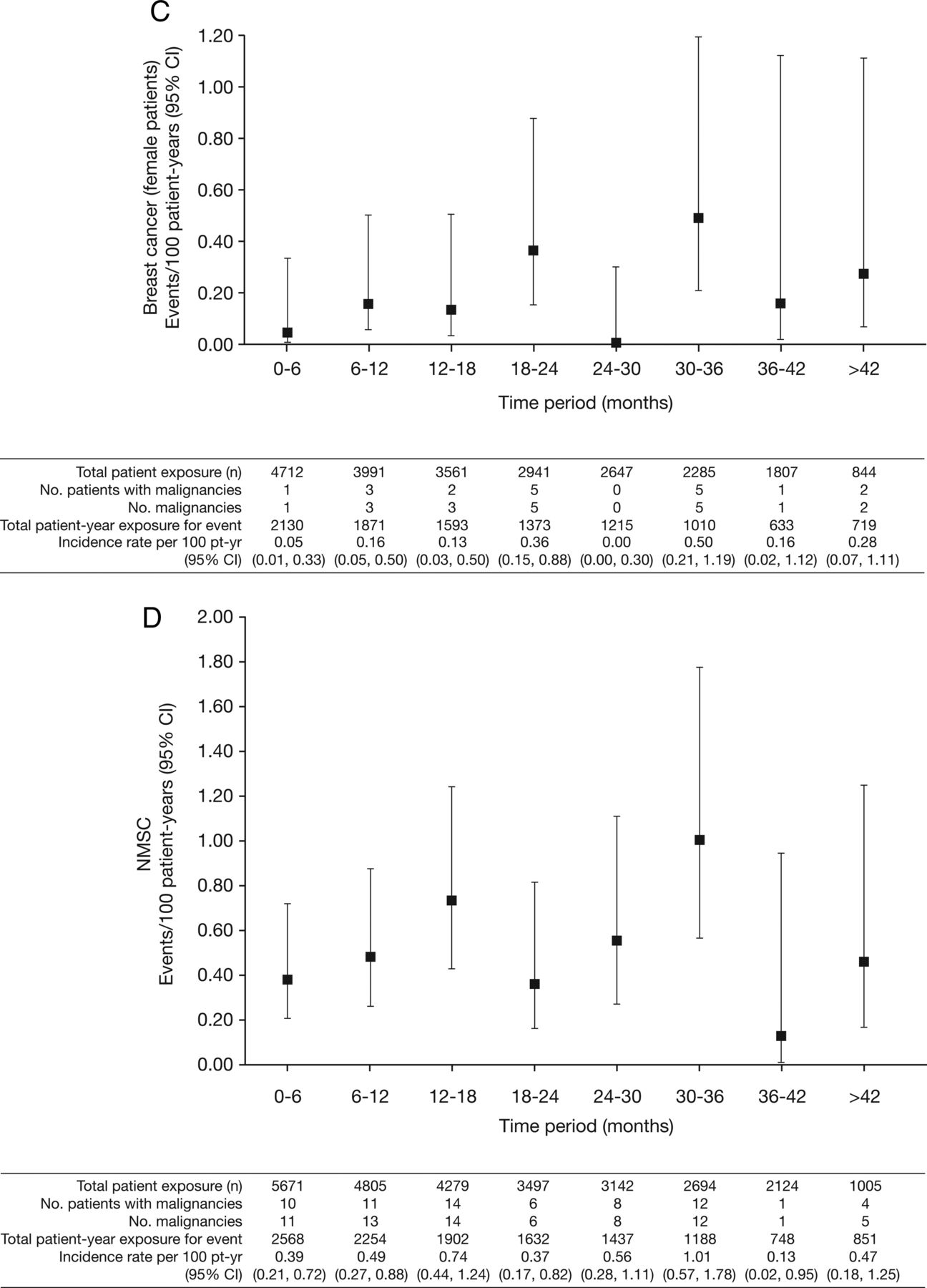
Incidence rates (95% CI) per 6-month intervals for (A) all malignancies (excluding non-melanoma skin cancer (NMSC)); (B) lung cancer; (C) breast cancer (female patients); (D) NMSC and (E) age-adjusted and sex-adjusted standardised incidence ratios (95% CI) per 6-month intervals for all malignancies (excluding NMSC). pt-yr, patient-years; SEER, Surveillance Epidemiology and End Result; SIR, standardised incidence ratio as compared with SEER database.
All malignancies (excluding NMSC)
In total, 107 of 5671 patients had malignancies (excluding NMSC); the overall IR for all tofacitinib-treated patients in the phase II, III and LTE studies was 0.85 (95% CI 0.70 to 1.02) events per 100 py (figure 1A). Total py exposure for all malignancies (excluding NMSC) is shown in figure 1A. In the phase III populations, malignancy IRs in the monotherapy and combination therapy groups (both doses) were 0.32 (95% CI 0.10 to 0.99) and 0.83 (95% CI 0.53 to 1.28); IRs in the tofacitinib 5 and 10 mg twice daily groups were 0.55 (95% CI 0.27 to 1.09) and 0.87 (95% CI 0.50 to 1.49). There was no association between tofacitinib treatment duration and overall malignancy risk (figure 2A).
The overall age-adjusted and sex-adjusted SIR for all malignancies (excluding NMSC) versus SEER among tofacitinib-treated patients was 1.17 (95% CI 0.96 to 1.41; table 2). SIRs were stable over time (figure 2E).
IRs and SIRs for all malignancies (excluding NMSC), lymphoma, lung and breast cancer for tofacitinib-treated patients with RA (across phase II, III and LTE studies)
Lung cancer
Twenty-four patients (12 men; 12 women) reported lung cancer: most cases were NSCLC; five patients were diagnosed with lung cancer within 6 months of receiving tofacitinib; 14/24 patients were current smokers, 6 were ex-smokers and 4 were non-smokers. The overall (phase II, III and LTE studies) IR in tofacitinib-treated patients was 0.19 (95% CI 0.13 to 0.28) events per 100 py (figure 1B). IRs for lung cancer observed in the phase III and LTE studies are presented in figure 1B. In the phase III studies, the IR for lung cancer in patients receiving tofacitinib 5 and 10 mg monotherapy was 0.00 (95% CI 0.00 to 0.82) and 0.21 (95% CI 0.03 to 1.52), respectively, and was 0.30 (95% CI 0.10 to 0.92) and 0.00 (95% CI 0.00 to 0.36) for patients receiving 5 and 10 mg tofacitinib in combination with DMARD therapy, respectively. IRs for lung cancer over time are shown in figure 2B. The SIR for lung cancer across phase II, III and LTE studies was 2.19 (95% CI 1.39 to 3.29; table 2).
Breast cancer
Nineteen patients (all women) reported breast cancer; the overall (phase II, III and LTE studies) IR was 0.18 events per 100 py (95% CI 0.12 to 0.28) (figure 1C). IRs for breast cancer over time are shown in figure 2C. The SIR for breast cancer across phase II, III and LTE studies was 0.78 (95% CI 0.47 to 1.22; table 2).
Non-melanoma skin cancer
Eighty-two NMSC events were reported in 66/5671 tofacitinib-treated patients; there were 44 cases of basal cell carcinoma (n=39) and 38 cases of squamous cell carcinoma (n=33). The overall (phase II, III and LTE studies) IR was 0.53 events per 100 py (95% CI 0.41 to 0.67; figure 1D). In the LTE studies, the rates of NMSC were higher in the tofacitinib 10 mg twice daily group versus 5 mg twice daily group (figure 1D). IRs for NMSC over time are shown in figure 2D.
Lymphoma
Ten cases (five in the phase III and five in the LTE studies) of lymphoma were reported for patients receiving tofacitinib. There was no apparent pattern of lymphoma occurrence associated with tofacitinib dose or therapy duration. For completeness, 2 of 10 lymphoma cases were included in patients from the second year of the ongoing phase III study NCT01039688 (ORAL Start), with exposure estimates based on randomisation ratios in the ongoing (second year) portion of this study. The overall IR (phase II, III and LTE studies) was 0.08 events per 100 py (95% CI 0.04 to 0.14; figure 1E); estimated total of 13230.87 py. Age-adjusted and sex-adjusted SIR was 2.64 (95% CI 1.27 to 4.86; table 2). Sequential cuts of the tofacitinib clinical programme data were completed from 2011 to 2013 to evaluate SIRs for lymphoma over time (see online supplementary appendix figure S1). Details of the 10 lymphoma cases are summarised as follows: six patients were treated with chemotherapy; one patient had no treatment information reported; one patient's lymphoma diagnosis was made through autopsy report (no treatment given); one patient with low-grade non-Hodgkin's lymphoma refused to be treated with recommended chemotherapy (reported by the investigator to do well without chemotherapy); and one patient had resection of the salivary gland with no chemotherapy or radiotherapy (the patient recovered according to the investigator). The types of lymphoma reported were histopathologically heterogeneous. One case was unequivocally positive for Epstein–Barr virus (EBV), two were equivocal and five were negative. Table 3 and online supplementary appendix present further details.
Summary of lymphoma/lymphoproliferative disorder in phase II, III and LTE studies
Deaths due to malignancy
There were 18 deaths due to malignancies in tofacitinib-treated patients. Ten were due to lung cancer (n=3, 5 mg twice daily; n=7, 10 mg twice daily), one each due to breast, colon, gastric, ovarian, gallbladder, hepatic and rectosigmoid cancers (all 5 mg twice daily) and synovial sarcoma (10 mg twice daily).
Discussion
The overall IRs, SIRs and types of malignancies observed in the tofacitinib clinical programme remained stable over time with increasing tofacitinib exposure (figures 1 and 2 and online supplementary appendix figure 1). SIRs in tofacitinib-treated patients (referent to SEER) (table 2) were within the range expected for patients with moderate-to-severe RA.27–41 In this analysis, the most frequently reported malignancy in tofacitinib-treated patients was lung cancer and, of 24 patients with lung cancer, 20 were current or former smokers. Five patients were diagnosed with lung cancer within 6 months of tofacitinib start. Given the known growth rate of solid tumours, it is highly likely these cancers were pre-existent prior to tofacitinib therapy. The types of lung cancer observed (predominantly NSCLC) were consistent with the demographic characteristics of the patient population.28 Risk factors associated with race may be a consideration for gastric cancer; all five cases occurred in Japan, which has been described as having the highest gastric cancer rates worldwide.42
Context for the data described here may be gained by reviewing results previously reported in the literature: point estimates (IRs: events/100 py) (95% CI) for patients with RA receiving biological DMARDs ranged from 0.61 (0.45 to 0.80) to 1.87 (1.02 to 3.13) for all malignancies (excluding NMSC),27–35 0.05 (0.02 to 0.13) to 0.92 (0.67 to 0.12) for lymphoma,27–31 ,35 ,37 ,38 ,40 0.15 (0.08 to 0.27) to 0.23 (0.19 to 0.27) for lung cancer,27 ,28 ,35 and 0.08 (0.03 to 0.17) to 0.21 (0.17 to 0.26) for breast cancer.27 ,28 ,34 ,35 In this report, the corresponding IRs (events/100 py) (95% CI) for tofacitinib were 0.85 (95% CI 0.70 to 1.02) for all malignancies (excluding NMSC), 0.08 (0.04 to 0.14) for lymphoma, 0.19 (0.13 to 0.28) for lung cancer and 0.18 (0.12 to 0.28) for breast cancer. Further discussion on specific outcomes is provided as follows.
Increased risk for NMSC development has been reported in patients with RA, and in patients prescribed TNFi and prednisone;43–45 two reviews of malignancy in patients receiving TNFi reported 45–102% increased risk of NMSC.40 ,46 In LTE, a higher rate of NMSC was seen in patients treated with tofacitinib 10 mg twice daily than tofacitinib 5 mg twice daily, whereas rates were similar between doses in phase III where randomisation between doses was balanced (figure 1D). The rate of NMSC appeared stable with continued tofacitinib exposure (figure 2D).
Lymphoma is a risk associated with RA and with agents used for RA treatment that affect the immune system. It is unclear whether lymphoma risk is increased further by MTX or TNFi, though some studies have reported that DMARDs were not associated with lymphoma risk.47 ,48 The types of lymphomas reported in the tofacitinib clinical programme were consistent with lymphomas described in the RA and general populations.31 ,49 ,50 Patients with RA are at increased risk of developing lymphoma than the general population (consistent with the observed age-adjusted and sex-adjusted SIR of 2.64 for tofacitinib)—possibly related to immunosuppressive therapy and RA severity. Some lymphomas in patients with RA arise in B cells in association with latent EBV infection; some lymphomas regress with reduced immunosuppressive therapy.35 ,38 ,51–53 Baecklund et al47 ,48 concluded that high-level RA disease activity coupled with long disease duration is associated with greater lymphoma risk.
EBV staining was positive in one case of lymphoma and equivocal in two of eight lymphomas tested (two from patients receiving tofacitinib monotherapy). Increased risk of EBV-associated lymphoma has been associated with high tofacitinib blood concentrations in renal transplantation studies of tofacitinib, in which patients received tofacitinib in combination with corticosteroids and potent immunosuppressive agents, such as basiliximab and mycophenolate mofetil. Ongoing analysis of LTE studies and postmarketing vigilance will be important to understand whether a relationship exists between tofacitinib and EBV-associated lymphomas in patients with RA.
When evaluating risk of malignancy, factors such as a dose–response relationship and cumulative duration of use may be important. There was no apparent or consistent association between tofacitinib dose and risk of malignancy except for NMSC in the LTE. However, numerically higher rates for all malignancies (excluding NMSC), breast cancer and lymphoma were observed with the higher 10 mg twice daily dose in phase III, but not in LTE (figure 1). Also, we acknowledge that the most susceptible patients at highest risk of cancer who develop a malignancy early are excluded from further trial participation, leading to a less at-risk cohort over time. However, we found that rates were generally stable over time or were even numerically lower during the first year of exposure (figure 2). This phenomenon has been observed in RA studies with TNFi and other biological DMARDs,30 ,54 ,55 and it has been suggested that the lower rates might be an effect of recruitment bias of healthier patients.
Study limitations
Our analysis was limited by reliance on randomised, placebo-controlled and LTE trials that reflect a selected patient cohort (here, the exclusion of patients with any previous cancer). While patients’ general health was assessed, there were no specific screening procedures that would detect undiagnosed malignancies. Also, patients who developed a malignancy (see Methods section for details) were barred from further trial participation, making analyses of additional risk for second or recurrent malignancies impossible.
Exposure to placebo (266 py) and adalimumab (190 py) was short and yielded imprecise IRs, so comparison to these study arms should be made cautiously. Those data were included in this report mainly for completeness. In LTE, assignment to treatment (tofacitinib 5 or 10 mg twice daily) was not randomised but based on index study enrolment, protocol guidance and investigator discretion. This has resulted in greater length of exposure to tofacitinib 5 mg twice daily, whereas the extent (in py) was greater with tofacitinib 10 mg twice daily, making interpretation of any dose differences uncertain. Also, no formal statistical analysis was performed on the incidence of malignancies between treatment groups, so conclusions are based on descriptive analyses only. Moreover, due to the presumed extended latency periods of some malignancies, patients will continue to require continued observation, beyond the median 2.35 years of exposure to tofacitinib represented in this analysis, and the relationship between greater cumulative duration of tofacitinib use and malignancy warrants further study.
Conclusion
The overall rates and types of malignancies observed in the tofacitinib clinical programme remained stable over time with increasing tofacitinib exposure. Continued longer-term surveillance is necessary to further evaluate any potential malignancy risk during tofacitinib treatment.
Acknowledgments
We would like to thank the patients, investigators and study team who were involved in the studies (NCT01262118, NCT01484561, NCT01164579, NCT01359150, NCT00976599, NCT00413660, NCT00603512, NCT01059864, NCT00147498, NCT00550446, NCT00687193, NCT00814307, NCT00847613, NCT00960440, NCT00856544, NCT00853385, NCT00413699, NCT00661661, NCT01039688). We also extend our gratitude to Professor Xavier Mariette (Université Paris-Sud, Le Kremlin Bicêtre, France) for his expert guidance and input into the earlier drafts of this manuscript. Editorial support, under our guidance, was provided by Martin Goulding and Jason Gardner of Complete Medical Communications and was funded by Pfizer.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
Contributors JRC, EBL and IVK contributed to analysis and interpretation of the data, reporting and writing the manuscript. KK was involved in study conception and design, acquisition of data, analysis and interpretation of data, and reviewing the manuscript. JG was involved in data analysis, interpretation, reviewing/editing the manuscript. BB contributed to data interpretation, and manuscript editing/review/approval. KS, LW and RR have contributed to the planning, conduct and reporting of the work described in the manuscript as well as contributed to the planning, authorship and review of the manuscript. All authors approved the final draft of the manuscript for submission.
Funding Pfizer (10.13039/100004319).
Competing interests JRC reports grants and personal fees from Genentech, UCB, Janssen, CORRONA, Amgen, Pfizer, BMS, Crescendo, AbbVie, outside the submitted work. EBL has acted for a consultant for Pfizer during the conduct of the study; and acted as a consultant for Pfizer outside the submitted work. IVK, KK, JG, BB, KS, LW and RR are employees and shareholders of Pfizer.
Ethics approval The studies were conducted in compliance with the Declaration of Helsinki, International Conference on Harmonisation Good Clinical Practice Guidelines, and relevant local country regulations. Patients provided written, informed consent. The final protocol, amendments and consent documentation were reviewed and approved by the Institutional Review Board and Independent Ethics Committee of the investigational centres.
Provenance and peer review Not commissioned; externally peer reviewed.