Article Text

Abstract
Objective Axial spondyloarthritis (AxSpA) represents a group of inflammatory axial diseases that share common clinical and histopathological manifestations. Ankylosing spondylitis (AS) is the best characterised subset of AxSpA, and its genetic basis has been extensively investigated. Given that genome-wide association studies account for only 25% of AS heritability, the objective of this study was to discover rare, highly penetrant genetic variants in AxSpA pathogenesis using a well-characterised, multigenerational family.
Methods HLA-B*27 genotyping and exome sequencing was performed on DNA collected from available family members. Variant frequency was assessed by mining publically available datasets and using fragment analysis of unrelated AxSpA cases and unaffected controls. Gene expression was performed by qPCR, and protein expression was assessed by western blot analysis and immunofluorescence microscopy using patient-derived B-cell lines. Circular dichroism spectroscopy was performed to assess the impact of discovered variants on secondary structure.
Results This is the first report identifying two rare private familial variants in a multigenerational AxSpA family, an in-frame SEC16A deletion and an out-of-frame MAMDC4 deletion. Evidence suggests the causative mechanism for SEC16A appears to be a conformational change induced by deletion of three highly conserved amino acids from the intrinsically disordered Sec16A N-terminus and RNA-mediated decay for MAMDC4.
Conclusions The results suggest that it is the presence of rare syntenic SEC16A and MAMDC4 deletions that increases susceptibility to AxSpA in family members who carry the HLA-B*27 allele.
- Ankylosing Spondylitis
- Autoimmune Diseases
- Inflammation
- Spondyloarthritis
- Low Back Pain
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Spondyloarthritis (SpA) represents a group of inflammatory rheumatic diseases whose main clinical feature is inflammation of the axial spine.1 The focus of the current study is axial spondyloarthritis (AxSpA). Although the precise aetiology of SpA remains unknown, there is mounting evidence of complex interplay of genetic, environmental and immunological factors.1 Despite the success of genome-wide association studies (GWAS) in ankylosing spondylitis (AS), where 25 genetic loci have reached a level of significance, the genetic contribution identified only explains 25% of heritability.2–5 This ‘missing heritability’ is attributed, at least in part, to inherent limitations of GWAS studies, which primarily assess one type of genetic variation (ie, single nucleotide polymorphisms) in a case–control approach, and consequently limits searches to common variants. Rare variants, which might also contribute to SpA susceptibility, occur in approximately 1% of the population, and many are likely to be specific to ethnic groups, isolates or families.6
That SpA clearly clusters within families, evident from the occurrence of multiple affected individuals within a family7 ,8 and the high recurrence ratio among siblings,9 is a compelling reason to investigate the genetics in this setting. The objective of this study is to discover rare, highly penetrant pathogenic variants in the pathogenesis of AxSpA using a large, well-characterised, multigenerational family and exome sequencing of affected and unaffected family members.
Methods
Study population
A large Caucasian multiplex AxSpA family of North European Ancestry from Newfoundland, Canada, was identified in a university-based rheumatology clinic. All available family members were invited to participate in this study and were systematically assessed using a standardised protocol by an experienced rheumatologist (PR). The Spondyloarthritis Consortium of Canada provided clinical and radiographic data, as well as DNA samples for the replication cohorts.
B-cell lines
Acid citrate dextrose anticoagulated whole blood was collected from family members. Lymphoblastoid B-cell lines (BCL) were generated by Epstein–Barr transformation of peripheral blood B cells as described previously.10
Nucleic acid extraction
DNA was extracted from EDTA anticoagulated whole blood using a traditional salting-out method.11 BCL pellets were resuspended in 0.5 mL TRIzol (Life Technologies) and RNA extracted as per manufacturer's instructions and dissolved in Molecular Biology grade water. RNA quality and quantity measures are provided in the online supplementary methods.
HLA-B*27 testing
Targeted analysis of the HLA-B locus located on 6p21.3 was performed using a commercially available kit (LABType SSO HLA-B locus kit) on a Luminex 100/200 platform as per manufacturer's instructions (One Lambda).
Exome sequencing
Samples were sequenced targeting whole genome exons with an average coverage of 110× using Illumina HiSeq 2000. The mapping of reads was aligned using Burrows Wheeler Alignment V.0.7.10, and the genome analysis toolkit (GATK) V.1.1.28 was used to call variants against the reference genome. To reduce false positive calling, 40% support reads was used as a cut-off for alternative allele. Analysis was carried out to detect rare mutations that segregate only within the affected individuals. Annovar (April 2014 version) was used for variant annotation.
Fragment analysis
DNA was amplified using specific primers for SEC16A and MAMDC4 using a standard touchdown reaction on a GeneAmp PCR System 9700 (Applied Biosystems). PCR primers, PCR product preparation, capillary electrophoresis and results analysis are provided in the online supplementary methods.
Linkage analysis
A phased VCF file for the nuclear family (ie, II-1, II-2 and III-2) was obtained using the GATK software (V.3.3). VCFtools (V.0.1.12b) were used to calculate pairwise r2, D and D′ for the genetic variants identified on chromosome 9 from 138 000 000 to 141 000 000 base pairs (GRCh37) of the nuclear subfamily.12 This genomic region includes the two novel deletions in SEC16A and MAMDC4. Linkage disequilibrium (LD) between SEC16A and MAMDC4 in the general population was investigated using DistilLD Database13 and GLIDERS.14
Quantitative PCR
Real-time PCR was performed using TaqMan Gene Expression Assays for SEC16A (Hs_00389570_m1) and GAPDH (Hs_99999905_m1) from Life Technologies. Samples were tested as per manufacturer's instructions and run on a StepOnePlus (Applied Biosystems). Triplicate samples were analysed using the comparative threshold cycle (ΔΔCT) method and results normalised to GAPDH. Statistical analyses were performed by One-way analysis of variance (ANOVA) and unpaired t test with GraphPad Prism V.6.04.
Western blot
RIPA lysates were prepared from BCL as described previously.15 Protein determination was performed using a bicinchoninic acid protein assay kit according to manufacturer's instructions (Thermo Scientific). Primary and secondary antibodies as well as the imaging system used are indicated in the online supplementary methods. Each sample was expressed as a ratio to tubulin, then compared with the proband. Statistical analyses were performed using one-way ANOVA and unpaired t test with GraphPad Prism V.6.04.
Circular dichroism spectroscopy
Detailed information on protein expression and purification prior to circular dichroism (CD) spectroscopy is indicated in the online supplementary methods. CD spectra in the far-ultraviolet range were recorded using a Jasco-810 spectrapolarimeter. The temperature (25°C) was controlled, and the scanning speed of the instrument was set at 100 nm/min with normal sensitivity. A water-jacketed cell (light path=0.5 mm) was used, and spectra were collected between 190 and 260 nm. Baselines were established using the appropriate buffers, and 30 spectra were collected and averaged for each sample.
Results
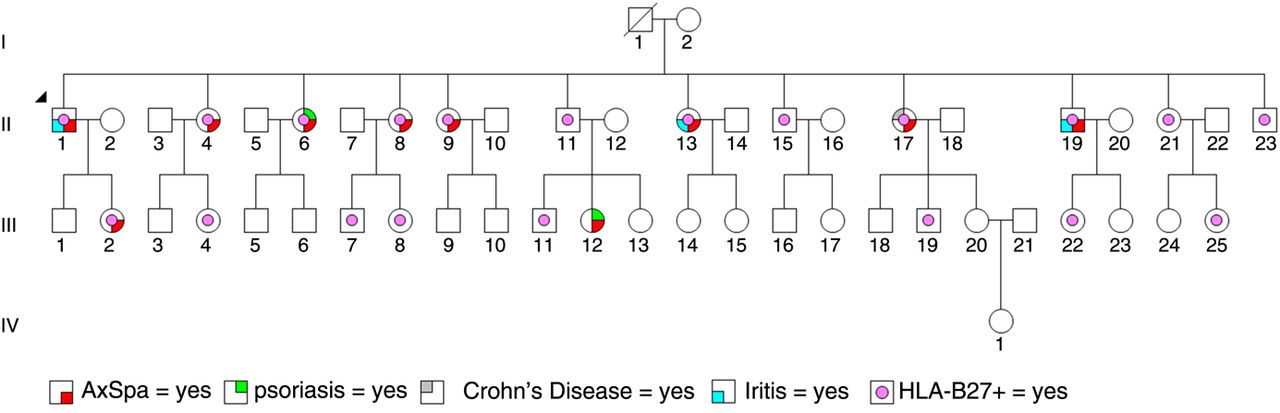
A well-characterised, multigenerational family with AxSpA was investigated to identify rare genetic variants (figure 1). All affected family members carried the HLA-B*27 allele (table 1). Exome sequencing was subsequently performed on selected family members with a minimum coverage of 110×. An exome sequencing analysis pipeline (see online supplementary figure S1) was used for the detection of genetic variants (see online supplementary table S1). Subsequent filtering and analysis revealed a 9 base pair in-frame deletion in SEC16A and a 20 base pair out-of-frame deletion in MAMDC4 (figure 2A), which segregated with AxSpA in the family (table 1). The chromosome 9 deletions located within exon 3 of SEC16A and exon 5 of MAMDC4 were confirmed bidirectionally using Sanger sequencing in family members primarily from generation II (figure 2B, C) with 7/9 of the clinically diagnosed AxSpA individuals carrying both deletions in synteny (table 1). In contrast, both deletions were not in synteny for all unaffected family members tested in generation II. Closer inspection revealed that family members diagnosed with AxSpA who carried both deletions in synteny in addition to the HLA-B*27 allele had an earlier age of symptom onset (20.2±3.14 vs 35.0±0; p=0.0074; t test with Welch's correction) compared with family members diagnosed with AxSpA who carried the HLA-B*27 allele but both deletions not in synteny.
Clinical information, genotype data (HLA-B27, SEC16A and MAMDC4) of the large, well-characterised, multigenerational family with numerous individuals clinically diagnosed with and without AxSpA

A large multigenerational family with several members clinically diagnosed with and without axial spondyloarthritis (AxSpA). Diagnosed comorbidities such as psoriasis, iritis and Crohn's disease are also indicated. The proband (II-1) in this family is indicated by an arrowhead.

Deletions located within SEC16A and MAMDC4. (A) Full exome sequencing analysis revealed a 9 base pair deletion located within SEC16A and a 20 base pair deletion located within MAMDC4 that segregated only within affected family members. Both deletions were confirmed using bidirectional Sanger sequencing of the proband (II-1). (B) The top panel is a representative chromatogram illustrating a portion of the wild-type sequence of SEC16A exon 3. The lower panel is a representative chromatogram illustrating a portion of SEC16A exon 3 with the in-frame deletion indicated by an arrow. (C) The top panel is a representative chromatogram illustrating a portion of the wild-type sequence of MAMDC4 exon 5. The lower panel is a representative chromatogram illustrating a portion of MAMDC4 exon 5 with the out-of-frame deletion indicated by an arrow.
The frequency of each deletion was assessed in publically available datasets to determine if either deletion represents a rare genomic event. Mining of the 1000 Genomes, National Heart, Lung, and Blood Institute, and in-house sequencing datasets (7849 total controls) revealed a frequency of 0.4% and 1.2% for the SEC16A and MAMDC4 deletion, respectively. To determine if either deletion represents a rare variant private to the study family, the frequency of the SEC16A and MAMDC4 deletion was assessed using fragment analysis in unrelated AxSpA cases and compared with unaffected controls (see online supplementary table S2). The SEC16A deletion produced a frequency of 0.87% in unrelated AxSpA cases compared with 0.84% in unaffected controls (p=0.925). Similarly, the MAMDC4 deletion produced a frequency of 1.38% in unrelated AxSpA cases compared with 1.41% in unaffected controls (p=0.926). Interestingly, both SEC16A and MAMDC4 deletions were only detected together in the AxSpA study family and were not detected together in over 900 unrelated AxSpA cases or in 1150 unaffected controls.
Linkage analysis, which was performed to estimate the degree of linkage between the SEC16A and MAMDC4 loci in the family, revealed that there is very strong LD (r2=1; D′=1) between the SEC16A and MAMDC4 loci within the nuclear subfamily (see online supplementary table S3; figure 3A, B). In contrast, SEC16A and MAMDC4 loci occur in separate LD blocks in the general population (see online supplementary table S4; figure 3C, D).

Linkage analysis was performed using VCFtools to estimate the degree of linkage between the SEC16A and MAMDC4 loci in the family and the general population. Analysis revealed that there is very strong linkage disequilibrium (LD) (r2=1; D′=1) between the SEC16A and MAMDC4 loci within the nuclear subfamily. The X and Y axes are genomic positions on chromosome 9, the deletions in SEC16A and MAMDC4 are located at positions 139370954 and 139748276, respectively. White represents a score of 0 and black represents a score of 1. Linkage disequilibrium cluster plots illustrating (A) r2 values and (B) the D′ values for the nuclear family. In contrast, (C) SEC16A and (D) MAMDC4 loci occur in distinctly separate LD blocks in the general population.
Gene and protein expression analysis was performed on select family members to determine if the SEC16A deletion affects gene transcription or translation. SEC16A gene expression was not significantly different in AxSpA individuals carrying the deletion compared with AxSpA individuals not carrying the deletion (p=0.2383; figure 4A). Western analysis revealed that Sec16A protein expression was not significantly different in AxSpA individuals carrying the deletion compared with AxSpA individuals not carrying the deletion (p=0.3849; figure 4B, C).
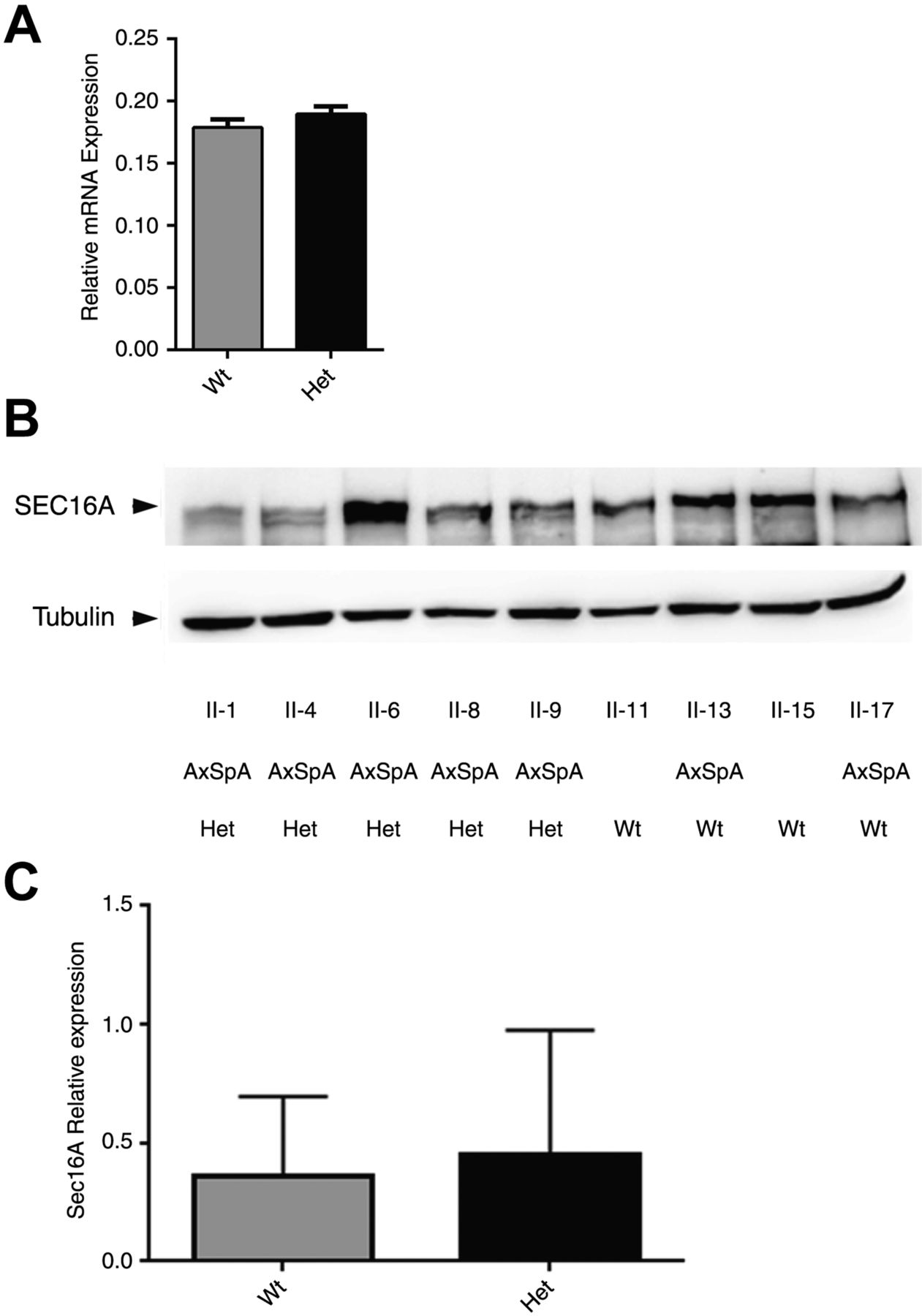
SEC16A gene and SEC16A protein expression is not altered among individuals with the deletion mutation. (A) The relative expression of SEC16A mRNA was determined by real-time quantitative PCR (qPCR) analysis of patient-derived B-cell lines. SEC16A relative expression was compared between family members with wild-type (Wt) and heterozygous (Het) genotypes for the SEC16A and MAMDC4 deletion. Results are expressed as mean±SEM and analysed by t test. (B) Sec16A protein expression of RIPA lysates from patient-derived B-cell lines of family members. Blots are representative of three separate experiments. Densitometric analysis was performed to determine the level of SEC16A expression relative to tubulin as a loading control. (C) Comparison of protein expression between family members with Wt and Het genotypes for the SEC16A deletion. Results are expressed as mean±SEM and analysed by t test.
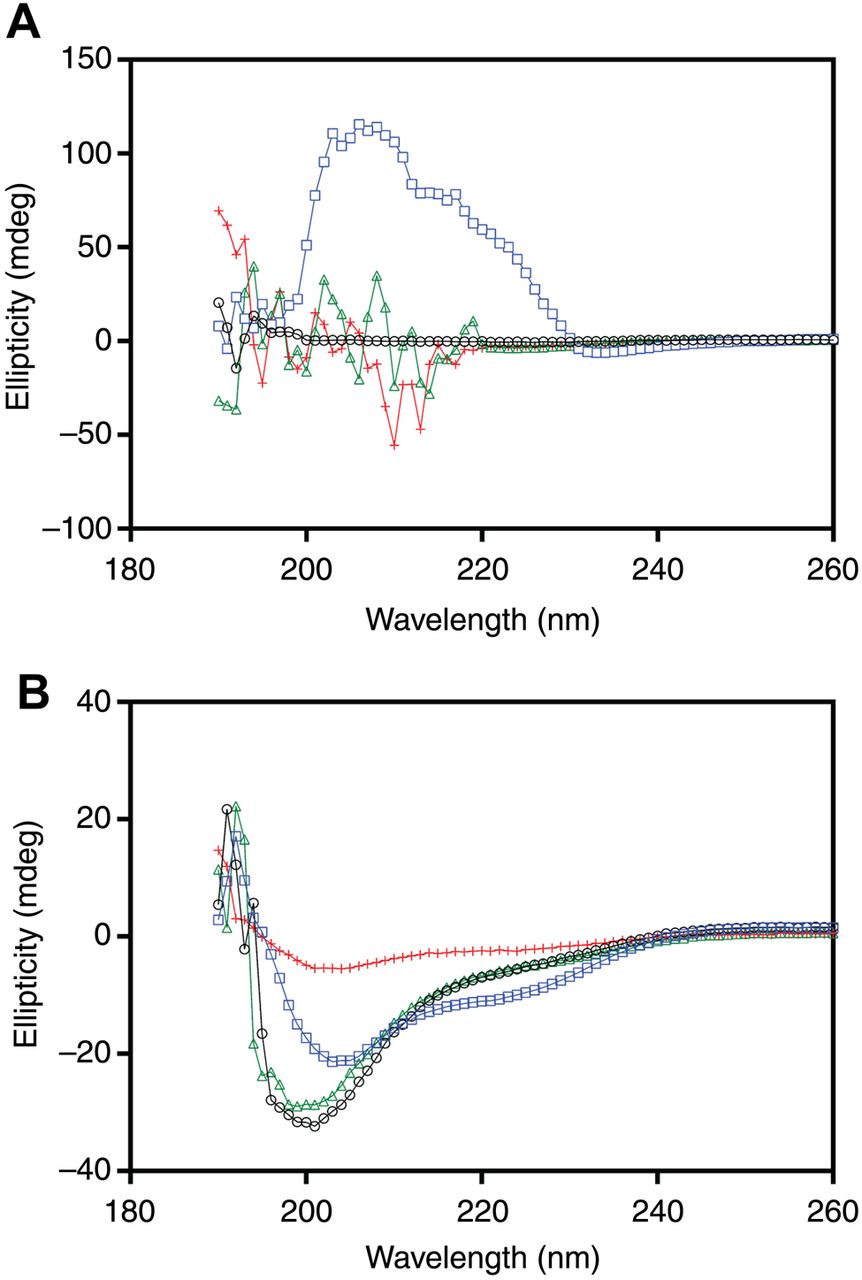
CD spectroscopy was performed to determine if the SEC16A deletion affects protein structure. Given that the SEC16A deletion is located in a region that encodes the N-terminus of Sec16A, a 40 amino acid region around the deletion was initially selected using PONDR-FIT. CD spectroscopy demonstrated that the wild-type and deletion peptides have a disordered secondary structure (figure 5A). In the presence of trifluoroethanol (TFE), the wild-type peptide adopted a secondary structure, whereas the peptide encoding the SEC16A deletion clearly lacked secondary structure. A disordered secondary structure was also confirmed using CD spectroscopy on the complete N-terminus peptide (figure 5B). In the presence of TFE, the wild-type full-length N-terminus adopted an alpha helical secondary structure; however, the full-length N-terminus encoding the SEC16A deletion (369–371) had a similar spectra profile but exhibited decreased signal amplitude.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Circular dichroism (CD) spectroscopy performed on the 40 amino acid peptide and the complete N-terminus peptide of Sec16A. (A) CD spectroscopy of the 40 amino acid peptides demonstrated that both the Sec16A wild-type (o; black) and mutant (Δ; green) peptides have a secondary structure that is disordered. The wild-type peptide (□; blue) in the presence of trifluoroethanol (TFE) adopted a distinct secondary structure (broad peak from 230 to 200 nm and the disappearance of the negative peak at 195 nm). The mutant peptide (+; red) in the presence of TFE failed to adopt a distinct secondary structure. (B) CD spectroscopy of the complete N-terminus also demonstrated that both the Sec16A wild-type (o; black) and mutant (Δ; green) peptides have a secondary structure that is disordered. The wild-type peptide (□; blue) in the presence of TFE adopted a distinct secondary structure, whereas the mutant peptide (+; red) in the presence of TFE had a spectra similar in shape to the wild-type peptide, but exhibited a decreased signal amplitude. Each CD spectroscopy graph is representative of the average of 30 spectra.
Finally, experiments were conducted to investigate if the SEC16A deletion disrupts co-localisation with Sec31A. Immunofluorescence microscopy using patient-derived BCLs from individuals with the SEC16A deletion demonstrated no qualitative change in Sec16A or Sec31A expression alone or in their co-localisation (see online supplementary figure S2).
Discussion
A well-characterised, multigenerational AxSpA family was investigated using exome sequencing and HLA-B*27 genotyping. Investigation was primarily limited to individuals within generation II as those individuals were available for precise clinical phenotyping. Individuals from generation III (except III-2) were not systematically assessed given their lack of availability to participate in the study, their relatively young age and the fact that their assignment of phenotype could change with time, which could confound data analysis. The perfect concordance of exome and Sanger sequencing excludes SEC16A and MAMDC4 deletions as technical artefacts. Both deletions were inherited maternally (I-2), whereas the HLA-B*27 allele was presumably inherited paternally (I-1). Furthermore, given that I-2 is HLA-B*27 negative and that all 12 siblings in generation II are HLA-B27 positive, it was deduced that I-1 was homozygous for the HLA-B27 allele.
That linkage analysis revealed that there is very strong LD (r2=1; D′=1) between the SEC16A and MAMDC4 loci within the nuclear subfamily and extended family members, whereas SEC16A and MAMDC4 loci occur in distinctly separate LD blocks in the general population, which suggests that a different genomic architecture is present on 9q34.3 in this family compared with the general population; a finding possibly attributed to a large intergenic deletion or a recombination event not detectable by exome sequencing.
That numerous variables affect genotype–phenotype correlation in complex disease including reduced penetrance, variable expressivity, epistatic interactions and gene–environment interactions, it was not surprising that a perfect genotype–phenotype correlation was not achieved in all family members within generation II. Interestingly, the discordant genotype–phenotype relationship occurred in two individuals (ie, II-13 and II-17) that had less severe disease and a significantly later age of onset. The apparent discrepancy in population frequency between publically available datasets and the replication cohort is largely attributed to differences in coverage depth as gaps in the public datasets result in detection bias.
SEC16A contains 32 exons and encodes a 250 kDa protein comprising 2357 amino acids. The deletion results in the removal of three amino acids (small for gestational age) within the N-terminus, producing a mildly truncated transcript. MAMDC4 contains 27 exons and encodes a 131 kDa protein (ie, endotubin) comprising 1216 amino acids. The deletion results in a premature termination codon (p.Val185Glyfs*2), producing a severely truncated transcript containing only 187 amino acids, which is likely rapidly degraded by nonsense-mediated RNA decay (NMD). The primary focus of this study was to increase our understanding of how the SEC16A deletion contributes to pathogenicity given that (1) Sec16A functions as a scaffold protein with multiple central roles for coat protein complex II (COPII) vesicle formation and trafficking,16–19 which may be of relevance to AxSpA pathogenesis; (2) a recent study in patients with inflammatory bowel disease (IBD) where exome sequencing identified multiple rare novel SEC16A variants;6 (3) very little is known regarding the structure and function of the MAMDC4 encoded protein;20 and (4) the MAMDC4 deletion is a target for NMD.
That the SEC16A deletion failed to alter gene or protein expression was not unexpected considering this deletion failed to produce a severely premature transcript therefore avoiding NMD, suggesting that a deleterious effect may be attributed to a mechanism independent of transcription or translation. Interestingly, the Sec16A N-terminus, which is quite disordered and possesses many evolutionary conserved disordered binding regions, is critical for COPII assembly.21 ,22 Results from CD spectroscopy using a 40 amino acid peptide of the Sec16A N-terminus (selected using PONDR-FIT23) with and without the deletion confirmed that both were intrinsically disordered. That TFE induced a distinct structure only with the wild-type peptide and not with the deletion peptide suggests that the deletion of three amino acids within a short peptide is sufficient to interfere with local secondary structure. CD spectroscopy of peptides comprising the complete N-terminus of Sec16A confirmed the results generated with the shorter peptide, and further indicated that the deletion is sufficient to interfere with secondary structure of the entire N-terminus. The decreased signal amplitude could result either from a perturbation in the structure of the deletion peptide or from a global destabilisation that produces an increased population of unfolded protein at equilibrium with a well-folded population.24 Therefore, the SEC16A deletion may alter the ability of Sec16A to adopt an ordered structure in the presence of its receptor proteins, which may contribute to AxSpA in this family.
That Sec16 serves as a template for the Sec13–Sec31 coat, that both Sec16 and Sec31 share an ACE1 element and that direct interactions between Sec16 and Sec31 facilitate COPII assembly,18 the effect of the SEC16A deletion on Sec31A co-localisation was investigated to better define the functional impact of the SEC16A deletion. That the SEC16A deletion was without effect on Sec31A co-localisation could suggest that a mechanism completely independent of Sec31A (eg, another component of the COPII coat) might be altered by the SEC16A-induced conformational change. For example, Sec31A may be recruited to the COPII assembly site, but the direct interaction with Sec16A might be impaired due to the deletion-induced conformational change. Future investigations using electron microscopy are warranted to address this possibly.
Unfortunately, studies investigating the dysfunction of Sec16A are lacking. However, a role for Sec16 in AxSpA pathogenesis may involve several possible mechanisms attributed to known interactions of Sec16A with nuclear factor kappa B,25 TRAF3,26 COPII-binding protein Bap3127 and the MAPK pathway.28 ,29 Any one or multiple interactions may be negatively disrupted by the SEC16A-induced conformational change.
This study is not without limitations. As exome sequencing does not interrogate the complete genome, other events occurring outside the coding region (eg, distal enhancer, silencers) might also contribute to disease susceptibility in the study family. For example, the 400 kb region between SEC16A and MAMDC4 contains an extremely large number of common variants of which many, if not most, are non-coding and are unlikely to have been picked up by the sequencing method used. Any of these variants could be the true disease-associated variant if in LD with the SEC16A and/or MAMDC4 variants. Although the study design with respect to exome coverage depth and variant filtering was very conservative, the possibility exists that a pathogenic variant could have been missed. Since the effect of the SEC16A deletion was investigated functionally using only B cells grown in culture, the contribution of other cell types and the effect of the SEC16A deletion in those cell types are unknown.
That the population frequency of SEC16A and MAMDC4 was low and not significantly different between unrelated AxSpA cases and controls, that both deletions were only detected together in the study family or in a large replication cohort (n=2050), that SEC16A and MAMDC4 are in strong LD within the family and that family members diagnosed with AxSpA who carried both deletions in synteny in addition to the HLA-B*27 allele had an earlier age of symptom onset suggest that it is the presence of the private rare deletions in synteny that increases susceptibility and modifies the disease course in the affected family members who carry the HLA-B*27 allele. Interestingly, that affected family members carried both SEC16A and MAMDC4 deletions and tested positive for HLA-B27 also raises the possibility that their encoded proteins are involved in HLA-B27 transport. We do acknowledge that the finding that AxSpA family members who carried both deletions in synteny in addition to the HLA-B*27 allele had an earlier age of symptom onset compared with AxSpA family members who carried the HLA-B*27 allele but both deletions not in synteny is based on a very small number of patients, and so this genotype–phenotype correlation needs to be independently validated.
While this is the first report identifying potentially rare novel private pathogenic deletions in SEC16A and MAMDC4 in AxSpA, a study employing exome sequencing of patients with IBD identified rare novel SEC16A variants. Specifically, a non-synonymous rare SEC16A variant (c.1480G>C; p.G494R) predicted to be pathogenic was identified.6 Interestingly, that variant was located in exon 3, the same exon as the deletion discovered in this study. The finding of rare pathogenic SEC16A variants in a related autoimmune disease and the conformational change induced by deletion of three highly conserved amino acid residues from the intrinsically disordered and critically important N-terminus of Sec16A in the current study supports a pathogenic role of SEC16A in AxSpA pathogenesis in this family. That MAMDC1, which has been identified as a candidate gene for systemic lupus erythematosus,30 is a paralog of MAMDC4, and that the MAMDC4 deletion is predicted to be pathogenic through NMD, is consistent with pathogenicity. However, a better understanding of the effect of the MAMDC4 deletion on structure and function of endotubin warrants further investigation. In conclusion, this study highlights the importance of private rare variants in AS/AxSpA and suggests that a proportion of the heritability unaccounted for by common variation may reside with higher risk rare variants, and even private variants.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
- Data supplement 2 - Online figures
- Data supplement 3 - Online tables
Footnotes
Handling editor Tore K Kvien
Contributors DDO, MU, NH, RI and PR designed the study. MU, DC, MH, AAM and WL collected the data. NH, RI and PR provided clinical information and samples. DDO, MU, DC, LP-C, JZ, SMMH, LP-C and PR analysed the data and critically interpreted the results. DDO prepared the first version of the manuscript. All the coauthors reviewed the draft versions and gave their approval of the final version of the manuscript.
Funding Atlantic Canada Opportunity Agency—Atlantic Innovation Fund (ACOA-AIF) (grant number 781-19095-199318), Canadian Institute of Health Research (CIHR) (grant number FRN 127002), Research and Development Corporation (RDC) (grant number 5404.1162.104), Newfoundland and Labrador, and Spondyloarthritis Consortium of Canada (SPARCC).
Competing interests None.
Patient consent Obtained.
Ethics approval The ethics committees from all participating hospitals have approved the study (HREA#1999.172).
Provenance and peer review Not commissioned; externally peer reviewed.