Article Text

Abstract
Objective To investigate the possible occurrence of early thymic failure and premature senescence of naïve and memory T-cells in patients with axial spondyloarthritis (aSpA).
Methods Prospective, cross-sectional study of consecutive patients with aSpA (n=51), rheumatoid arthritis (RA, n=51) and healthy controls (HCs, n=50). Demographic, clinical and laboratory parameters were collected in all patients and we isolated naïve (CD45RA+) and memory (CD45RO+) CD4+ and CD8+ T-cell subsets by MACS technology. T-cell receptor rearrangement excision circle (TREC) and telomere length were measured by real-time PCR. We used TRECs as a surrogate for thymus function and telomere length as an indicator of cellular senescence. Telomerase activity was analysed with the Telomeric Repeat Amplification Protocols.
Results We observed a premature decline of thymic output in patients with aSpA and patients with RA compared with HCs as indicated by a reduction of TREC levels in naive T-cells (aSpA: age adjusted regression coefficient (regcoeff) for CD4+CD45RA+ T-cells −2.566, p=0.023; RA regcoeff=−2.844, p=0.008). Telomere length of all CD4+ and CD8+ T-cell subsets was reduced in young patients with aSpA compared with HCs, whereas data for patients with RA were comparable with HCs. Telomerase activity was inversely correlated with telomere length in HCs (correlation coefficient (corcoeff)=−0.532, p<0.001) but not in patients with aSpA (corcoeff=−0.056, p=0.697) and RA (corcoeff=−0.003, p=0.982).
Conclusions Our data indicate an age-inappropriate shrinkage of thymic output, an inappropriate shortening of telomeres in young patients with aSpA and an impaired telomerase enzyme in patients with aSpA and RA.
- T Cells
- Spondyloarthritis
- Autoimmune Diseases
- Rheumatoid Arthritis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Clinical and experimental data suggest a central role of T cells in the pathogenesis of axial spondyloarthritis (aSpA). Particularly, the strong linkage between human leucocyte antigen (HLA)-B27 and aSpA indicates that cytotoxic T-cell responses may play a key role.1 Our previous observation of an accumulation of senescent CD4+ T cells in patients with ankylosing spondylitis (AS) further suggests that CD4+ T cells contribute to the evolution of the disease.2 Senescent CD4+ T cells are characterised by the loss of the costimulatory molecule CD28, a TH1-type polarisation and the ability to lyse target cells. In AS, CD4+CD28− T cells were linked with worse clinical outcomes,2–4 and in other immune-mediated diseases and the general population, CD28− T cells and the contraction of lymphocytic telomeres, another sign of T-cell senescence, were valuable biomarkers for an inefficient vaccine response, cardiovascular events, malignancy and mortality.5–7
Whether immunosenescence is also one of the factors causing aSpA, and which mechanisms trigger the accumulation of senescent T-cells in AS patients, are questions that are still poorly understood. In rheumatoid arthritis (RA), early T-cell ageing was explained by premature shrinkage of thymic output resulting in accelerated homeostatic proliferation of existing T cells.8 Consequently, telomeres are progressively lost leading to replicative senescence. In healthy individuals, T cells may induce telomerase after their activation to restore telomeres, whereas in RA this enzyme is defective undermining homeostatic control of the naïve T-cell compartment.9
Whether early loss of thymus function, inappropriate telomere shortening of naïve and memory T-cells as well as telomerase insufficiency also occur in aSpA has been addressed by the present study.
Methods
Study population
We prospectively recruited 51 consecutive patients with aSpA fulfilling the Assessment in Spondyloarthritis International Society classification criteria,10 ,11 51 patients with RA according to the 2010 American College of Rheumatology/European League Against Rheumatism criteria12 and 50 healthy controls (HCs). We defined AS according to the modified New York criteria.13 There was neither evidence of chronic infections nor malignant disease in any individual as determined by history, clinical and routine laboratory examinations. This study was approved by the Institutional Review board of the Medical University Graz and written informed consent was obtained from each individual.
All patients underwent full medical history (including review of clinical records regarding a history of inflammatory bowel disease (IBD), uveitis and/or psoriasis) and clinical examination recording the number of tender (TJ) and swollen joints (SJ). Patients’ global assessment of disease activity (PGA), patients’ pain assessment (Ptpain) and evaluators’ global assessment (EGA) were determined on visual analogue scales (range 0–100 mm). Blood samples were routinely tested for erythrocyte sedimentation rate (ESR, range 0–10 mm/first hour) and C-reactive protein (CRP; range 0–5 mg/L) levels. Patients with aSpA were assessed using the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI),14 the Bath Ankylosing Spondylitis Functional Index (BASFI)15 and the Bath Ankylosing Spondylitis Metrology Index (BASMI),16 and patients with RA were assessed using the simplified disease activity index (SDAI)17 and the Disease Activity Score 28 (DAS28).18
Flow cytometry
FACS analysis of T-cell subsets was performed according to a routine protocol. In brief, erythrocytes were lysed and cells were incubated with antibodies against CD3 (Becton Dickinson, San Diego, USA), CD4, CD8, CD28, CD45 (all Beckman Coulter, Brea; USA), CD45RA (Becton Dickinson) and CD45RO (Beckmann Coulter). For intracellular staining of Ki-67 (Becton Dickinson), cells were permeabilised with IntraPrep Reagents (Beckman Coulter) and measured within 2 h. All analyses were performed using Cytomics FC500 (Beckmann Coulter) and using CxP software.
Isolation of T-cell subsets
Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll density gradient centrifugation as previously described.2 CD4+ and CD8+ T cells were obtained using negative selection MACS-kits according to manufacturer's instructions (CD4+ T-Cell Isolation Kit and CD8+ T-Cell Isolation Kit; Miltenyi, Bergisch Gladbach, Germany). In a second step, CD45RA+ and CD45RO+ T cells were obtained via positive selection using CD45RA or CD45RO Microbeads (Miltenyi).
Quantification of T-cell receptor rearrangement excision circles
T-cell receptor excision circles (TRECs) from PBMCs, naïve and memory CD4+ and CD8+ T-cells were analysed as described previously.19 In brief, DNA was extracted using QIAamp DNA Mini Kit (Qiagen, Chatsworth, California, USA). TREC concentrations were determined by quantitative real-time PCR based on the coding TREC sequence using LightCycler 480 II (Roche) and primers to amplify a DNA fragment 108 bp across the remaining recombination sequence δrec/ψα (5′-CACATCCCTTT CAACCATGCT-3′ and 5′-GGTGCAGGTGCCTATGC-3′). For quantification, a synthetic internal standard (Ingenetix, Vienna, Austria) was used. PCR reaction ran with 0.25 μg DNA, primers and hybridisation-probe labelled with FAM-TAMRA (5′-ACAC CTCTGGTTTTTGTA AAGGTGCCCAC-3′). To avoid bias by different numbers of naïve T cells, TRECs were calculated in relation to numbers of T cells.
Determination of telomere length
We measured telomere lengths using DNA from PBMCs, isolated naïve and memory CD4+ and CD8+ T-cell subsets by quantitative real-time PCR analysis and LightCycler FastStart DNA Master SYBR Green I (Roche) according to manufacturer's instructions. The following primers were used: 5′-CGGTTTGTTTGG GTTTGGGTTTGGGTTTGGGTTTGG GTT-3′ and 5′-GGCTTGCCTTACCCTTACCCT TACCCTTAC CCTTACCCT-3′. A single copy gene (36B4) (fw primer: 5′-CCCATT CTATCATCAACGGGTACAA-3′; rev primer: 5′-CAGCAAGTGGGAAGGTGTAATCC-3′) served for normalisation. Ct-values of test samples and control single copy genes were calculated and 2ΔCt value was determined.20
Telomerase assay
For determination of telomerase activity naïve and memory CD4+ and CD8+ T-cells were isolated and cultured in advanced RPMI 1640 (GIBCO) in the presence of 30 U/mL rIL-2 (Sigma, Vienna, Austria) and CD3/CD28 Dynabeads (bead:cell ratio 1:1; Invitrogen). After 3 and 6 days, 2 × 105 cells were harvested and telomerase activity was determined using TeloTAGGG Telomerase PCR ELISAplus kit (Roche) according to ‘Telomeric Repeat Amplification Protocols (TRAP)’.21
Statistical analysis
All statistical analyses were performed using SPSS, V.20.0 (Chicago, Illinois, USA). For data with normal distribution (according to Kolmogorov–Smirnov test), the mean (±SD) is shown and we applied the Student's t tests for comparisons. Otherwise, we indicated the median (range) and used the Mann–Whitney U test. Bonferroni's adjustment was applied in case of multiple testing (as stated).
We included consecutive samples of patients with aSpA and RA in our study to produce data that are generalisable to patients with aSpA and RA visited in clinical routine rather than using matched samples. Therefore, we adjusted for differences regarding age and sex by statistical means: First, we divided patients into five age groups: I: 21–35 years, II: 35.1–45, III: 45.1–55, IV: 55.1–65 and V: >65 years (see online supplementary table S1 for numbers and the mean of age in each group). Second, we performed regression analyses adjusting for age and other possible confounders as outlined below.
The following regression models were conducted: (a) linear inclusive multivariate regressions (method of least squares) were carried out to investigate a possible association between the primary diagnosis and TRECs, telomere lengths and telomerase activity in PBMCs, naïve and memory CD4+ or CD8+ T-cells (dependent variables) adjusting for age, sex and Ki-67+ cells. Ki-67 was included to account for the number of proliferating cells as both, TRECs and telomere length are reduced by cell proliferation.19 An interaction term age*aSpA and age*RA was introduced into the models to address specific differences between patients’ groups according to age; (b) we performed multiple regression models for patients with aSpA and RA exploring the possible influence of clinical parameters (BASDAI, BASFI, BASMI, AS, IBD, peripheral arthritis or uveitis in patients with aSpA and number of TJ, SJ, EGA, PGA, Visual Analogue Scale (VAS), SDAI/DAS28 in patients with RA), current therapy (biologics, glucocorticoids), disease duration, laboratory markers (HLA-B27 in aSpA, rheumatoid factor (RF)/anticyclic citrullinated peptide (anti-CCP) antibodies in RA as well as ESR and CRP) on TRECs, telomeres and telomerase activity in PBMCs and T-cell subsets. All models were corrected for age because of the strong influence of this factor on the dependent variables. A multivariate inclusive regression model was projected in case any ≥2 variables were significantly associated with the dependent variable.
We used the logarithmic transformation for the dependent variables because of their non-parametric distribution. CRP, RF and anti-CCP were categorised (normal/abnormal) because of high skew to the left. Normality, linearity and homoscedasticity of all regression models were tested and significant colinearity of predictors was excluded. For sensitivity analysis we excluded high leverage cases and cases producing low/high DFBETAs and/or large Cook values.
Results
Patients’ characteristics
Clinical characteristics are depicted in table 1. As expected, patients with aSpA were younger and more commonly men than patients with RA and HCs. Fifteen (29.4%) patients with aSpA had active disease as indicated by a BASDAI ≥4, and 17 (33.3%) patients had a disease duration ≤2 years. In the RA cohort, 9 (20.9%), 14 (32.6) and 12 (27.9) out of the 43 patients with available SDAI values had high, moderate or low disease activity, respectively, and 8 (18.6) patients were in remission.22 Twenty-three (45.1%) patients with RA had early disease (≤2 years’ duration).
Patients’ characteristics
Reduced thymic function in young patients with aSpA
We compared thymus function between patients with aSpA and RA as well as HCs by the determination of TRECs. TRECs are small stable DNA episomes that are formed during T-cell receptor rearrangement in the thymus. In periphery, TRECS are concentrated in naïve T-cells and are subsequently diluted by homeostatic proliferation of T cells compensating for thymic failure.
As depicted in table 2, we found a striking premature decline of TRECs in CD4+CD45RA+ T cells in young patients with aSpA as compared with HCs, whereas after the age of 45 years, results were comparable between groups. The reduction of TREC concentrations was similar in patients with aSpA and RA. We also observed lower TREC levels in PBMCs and naïve CD8+ T cells from patients with aSpA and RA compared with HCs.
Premature reduction of T-cell receptor rearrangement excision circle (TREC) levels in aSpA.
Multivariate regression analysis confirmed the observations reported above revealing significant differences between patients with aSpA or RA and HCs after adjustment for age, sex and percentages of proliferating cells. The interactions between diagnosis and age suggested a greater difference of TRECs between groups at younger than at older ages (table 3). Sensitivity analyses as described in the Methods section did not alter the primary result.
Multivariate regression analysis for TREC levels
Next, we tested whether TREC levels are linked with clinical variables or aSpA subtypes (see online supplementary table S2A). We did not include the presence or absence of psoriasis in regression analysis because only a single patient had this disease (see table 1). We observed a positive association between disease duration and TREC levels in PBMCs, naïve CD4+ and CD8+ T cells in patients with aSpA but not in patients with RA. Correction of the p value for multiple testing (since several regression models were conducted), however, resulted in an insignificant result (p=0.12–0.15). In RA, the presence of anti-CCP antibodies was linked to frequencies of TRECs in naïve CD4+ and CD8+ T cells (see online supplementary table S2B); however, also this result was not robust against Bonferroni's correction for multiple testing.
Reduced percentages of CD45RA+ T-cells in patients with aSpA
To investigate whether the shrinkage of thmyic function resulted in an altered composition of the peripheral T-cell pool we investigated the prevalence of circulating T cell subsets in patients and controls. As detailed in Supplementary Table S3, we observed a lower frequency of CD4+CD45RA+ T cells in patients with aSpA and RA compared with HCs, whereas the prevalences of circulating memory T-cell levels were increased.
Increased replicative history of naïve and memory T-cells in patients with SpA
Next, we tested whether thymic failure in patients with aSpA and RA is associated with premature reduction of telomere length. Data are shown in table 4. We observed shorter telomeres in PBMCs and T-cell subsets from patients with aSpA and RA compared with HCs among donors ≤35 years of age. The differences between patients with RA and HCs, however, did not reach statistical significance. In older patients, telomere lengths were comparable between patients and HCs except for a trend towards shorter telomeres in patients with aSpA of the age group of 35–45 years.
Telomere lengths are prematurely reduced in naïve CD4+ T cells of patients with aSpA and RA
Multivariate regression analyses correcting for age, sex and proliferating cells confirmed the differences concerning telomere lengths between patients with aSpA and HCs, and further revealed an interaction between age and aSpA diagnosis indicating greater differences between groups at younger age (table 5).
Multivariate regression analysis for telomere lengths
Concerning clinical variables, regression analysis revealed a significant association between DAS-28 and telomere lengths of PBMCs and all T-cell subsets in patients with RA, as well as associations between telomere lengths of memory CD8+, memory CD4+ and naïve CD8+ cells with disease duration, presence of AS and peripheral arthritis/HLA-B27, respectively, in patients with aSpA. None of these findings, however, was robust against adjustment for multiple testing (see online supplementary table S4A, B).
Telomerase assay
In healthy individuals, critical shortening of telomeres is prevented by the induction of telomerase enzyme. To investigate whether telomerase insufficiency contributes to the shortening of telomeres in patients with aSpA and RA, we determined telomerase by TRAP assay. Overall, we found an impaired induction of telomerase in CD4+CD45RA+ T-cells from patients with RA (results not stratified by age), whereas telomerase activity of this T-cell subset was normal in aSpA (see online supplementary table S5).
Correlating telomerase activity with age, we observed that naïve CD4+ T cells from HCs older than 45 years had a sixfold higher telomerase activity than younger individuals. In contrast, the telomerase activity diminished along with age in patients with aSpA and RA. Similar observations were made in the other T-cell subsets. Correlating telomerase activity with telomere length, we noted an increase of telomerase activity along with the shortening of telomeres in HCs, whereas this association was not found in patients with aSpA and RA (figure 1).

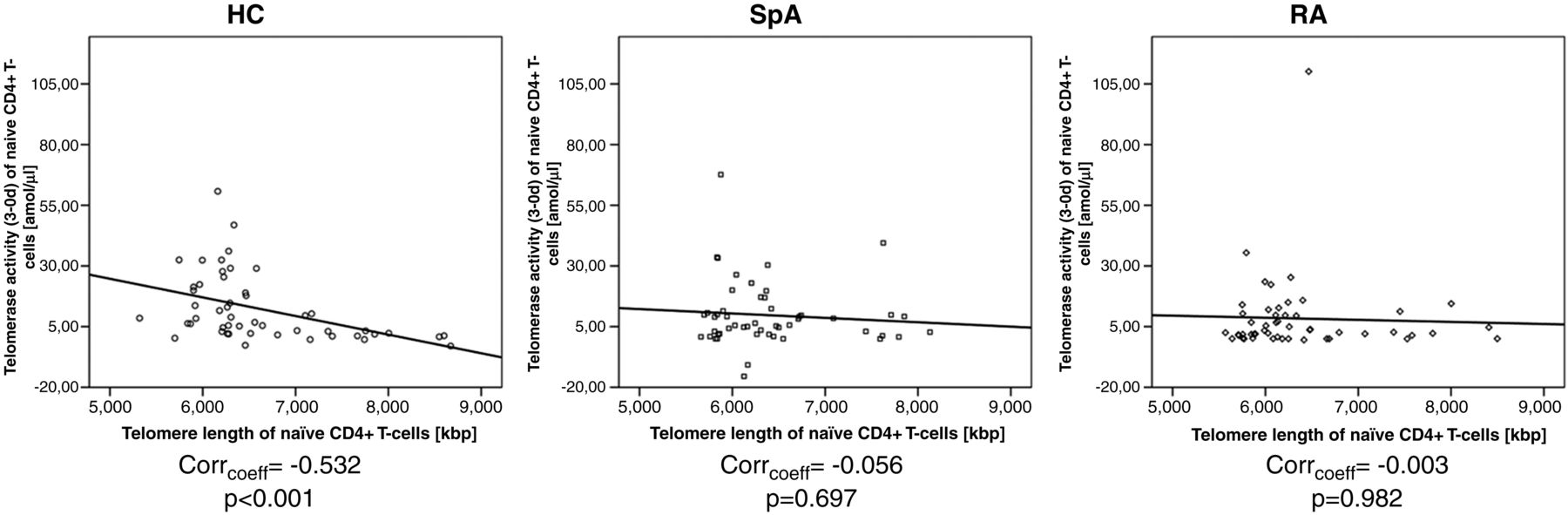{kind=link}
Lack of correlation of telomerase activity with telomere length in in patients with axial spondyloarthritis (aSpA) and patients with rheumatoid arthritis (RA). Graphs show correlation of telomerase activity (in amol/μ) with telomere length (in kbp) of CD4+CD45RA+ T-cells of patients with SpA and RA as well as healthy controls (HCs). Correlation coefficient and p values are given below the graph.
Using regression analysis, we found no significant association between telomerase activity in CD4+ T cells and clinical or laboratory variables in patients with aSpA and RA. Telomerase activity in naïve CD8+ T cells from patients with RA was linked with anti-CCP antibodies and in patients with aSpA, the telomerase activity of CD8+CD45RO+ T-cells was associated with the presence of IBD. Again, none of these findings was robust against adjustment for multiple testing (see online supplementary table S6A, B).
Discussion
The most important finding of this study is the dramatic reduction of thymic output in young patients with aSpA. This failure of thymus to produce new T cells may lead to the increased homeostatic pressure of existing cells triggering the observed progressive loss of telomeres and ultimately leading to premature T-cell senescence and accumulation of end-stage differentiated T cells.23 This concept is similar to that observed earlier for RA.8
The most critical question, whether these changes precede (or even cause) clinical onset of aSpA or are the consequence of a genuine disturbance of the immune system during (early) disease process, can only be speculated about. As thymic output and telomere lengths were similarly impaired in aSpA and RA, it is conceivable that the disturbance of T-cell homeostasis is a consequence rather than a cause of immune-mediated inflammation. Unfortunately, we were unable to investigate patients with aSpA at the preclinical stage to directly answer this question. The fact that we found no (robust) associations between immunological changes and clinical factors (including disease activity, disease subset, disease duration and treatment) argues against a prominent effect of chronic persistent aSpA on T-cell homeostasis.8 On the other hand, our study was of limited power to test the associations between clinical and immunological parameters thus bearing the possibility of a type II error.
From previous RA studies, we have learned that genetic factors and changes in the haematopoetic stem cell may contribute to early immune ageing.24 Healthy individuals carrying the HLA-DR4 epitope, the most important genetic risk factor for RA, yielded an age-inappropriate shortening of telomeres in naïve T cells.25 The reduction of telomere length in our young RA cohort, however, did not reach statistical significance, probably because of the low number of cases in this group.
Interestingly, we did not observe any differences between HLA-B27+ and HLA-B27− patients with aSpA regarding thymic function or telomere length but this does not exclude possible differences between HLA-B27+ and HLA-B27− healthy individuals and/or the linkage of early immune ageing with other polymorphisms associated with aSpA.26 We did not test for the presence of HLA-B27 in our HCs because of an inadequate power of our study for such comparisons (given an occurrence of HLA-B27 in 5%–10% of the healthy population).27 A future study in healthy individuals focusing on the association between genetic risk factors for aSpA and immunosenescence may be useful to better understand the mechanisms driving T-cell aging in aSpA.
In healthy individuals, telomerase partially restores telomere length of T-cells undergoing homeostatic or activation-induced proliferation, particularly in individuals at advanced age.28 We observed an inverse correlation between telomere length and telomerase activity in controls whereas this association was absent in both, patients with aSpA and RA. These findings may indicate an inappropriate low enzyme induction in these patients given that immunopathogenesis of aSpA and RA led to (or was caused by) premature erosion of telomeres. The exact mechanisms underlying a possible insufficiency of telomerase enzyme and/or additional factors contributing to shortened telomeres in patients with aSpA are unknown and should be clarified by future studies. In RA, an insufficient induction of the telomerase component human telomerase reverse transcriptase (hTERT) was observed suggesting a defect in transcriptional control of the hTERT gene.29 In patients with congenital dyskeratosis, mutations in proteins assembling and/or stabilising the telomerase complex were observed resulting in telomere shortening despite normal activity of the enzyme.29 ,30 An undiscovered polymorphism (or epigenetic modification) of the telomerase gene and/or an unstable telomerase protein complex may also reduce the efficacy of the enzyme to extend telomeres in aSpA.
Our study has a few limitations: First, the quantification of TRECs is an indirect measure of thymic function, although this method is generally accepted and supported by animal data.31 Direct assessment of thymic output is certainly preferable, however, for humans there is no such technique available. Although we adjusted our data for the number of proliferating cells (as indicated by the presence of Ki-67), we cannot completely rule out a bias introduced by naïve T-cell division.32 Our data should therefore be interpreted with caution. Second, patients with aSpA and RA were not matched for age, sex, disease duration, clinical activity and other factors. As aSpA predominately affects young men and RA, middle-aged women, such matching would produce artificial results for either of the two groups. Therefore, we chose to include consecutive patients from our outpatients’ clinic and to adjust for demographic differences by statistical methods. The fact, that results of our RA group were similar to the data of an earlier publication on RA strongly supports the validity of our findings.8
In summary, we observed an inadequate shrinkage of thymic output in patients with aSpA. Increased homeostatic proliferation of T-cells leads to premature shortening of telomeres that is not restored by telomerase enzyme.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
CD and MS contributed equally.
Contributors All authors contributed to conception and design of the study, acquisition and/or interpretation of data, drafting the article or revising it critically for important intellectual content and approved the final version of the manuscript. All authors had full access to all of the data (including statistical reports and tables) in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Funding This study was supported by the Oesterreichische Nationalbank (OeNB), Vienna (#13916 to M Schirmer), Innsbruck Medical University, Innsbruck.
Competing interests None.
Patient consent Obtained.
Ethics approval Institutional Review board of the Medical University Graz.
Provenance and peer review Not commissioned; externally peer reviewed.