Article Text

Abstract
Objectives Rheumatoid arthritis (RA)-specific anti-citrullinated protein/peptide antibodies (ACPAs) appear before disease onset and are associated with bone destruction. We aimed to dissect the role of ACPAs in osteoclast (OC) activation and to identify key cellular mediators in this process.
Methods Polyclonal ACPA were isolated from the synovial fluid (SF) and peripheral blood of patients with RA. Monoclonal ACPAs were isolated from single SF B-cells of patients with RA. OCs were developed from blood cell precursors with or without ACPAs. We analysed expression of citrullinated targets and peptidylarginine deiminases (PAD) enzymes by immunohistochemistry and cell supernatants by cytometric bead array. The effect of an anti-interleukin (IL)-8 neutralising antibody and a pan-PAD inhibitor was tested in the OC cultures. Monoclonal ACPAs were injected into mice and bone structure was analysed by micro-CT before and after CXCR1/2 blocking with reparixin.
Results Protein citrullination by PADs is essential for OC differentiation. Polyclonal ACPAs enhance OC differentiation through a PAD-dependent IL-8-mediated autocrine loop that is completely abolished by IL-8 neutralisation. Some, but not all, human monoclonal ACPAs derived from single SF B-cells of patients with RA and exhibiting distinct epitope specificities promote OC differentiation in cell cultures. Transfer of the monoclonal ACPAs into mice induced bone loss that was completely reversed by the IL-8 antagonist reparixin.
Conclusions While ACPA may induce OC activation, the conclusions concerning the specificity of these observations require additional experiments before detailed mechanisms can be elucidated. Further, it is also not yet clear if ACPA are pathogenetically involved in the initiation of the joint specific inflammation in ACPA-positive RA or not.
- Ant-CCP
- Autoantibodies
- Early Rheumatoid Arthritis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory joint disease. Anti-citrullinated protein/peptide antibodies (ACPAs) are found in the majority of patients with RA and are highly specific for RA.1 ACPAs comprise a collection of antibodies with different specificities towards citrullinated (cit)-epitopes. ACPAs may develop many years before the onset of joint inflammation,2 ,3 and their presence has been associated with bone loss.4 ,5
Citrullination is a post-translational modification in which arginine is converted to citrulline by an enzymatic reaction catalysed by peptidylarginine deiminases (PAD) in the presence of high levels of calcium.6–8 Citrullination was originally described as a physiological process in the terminal differentiation of the epidermis9–13 and during brain development,14 ,15 but it is also present in the context of inflammation.16 ,17
Bone resorption is a hallmark of RA, classically believed to reflect only the inflammatory burden in joints. Several pro-inflammatory cytokines present in the inflamed synovium, including interleukin (IL)-8,18 have been previously shown to stimulate osteoclasts (OCs).19 ,20 However, bone destruction may occur despite the disease being inactive21 and even in the absence of detectable inflammation in the joints of ACPA-positive individuals at risk of developing RA who do not yet have the disease.22 One potential explanation for these observations has been provided by the recent finding that ACPAs directed against mutated cit-vimentin and purified from serum of patients with RA could induce OC activation in vitro and bone resorption in vivo after transfer to mice.20 However, the molecular mechanisms and mediators involved in ACPA-induced OC activation are largely elusive. The aim of the present study was accordingly to dissect the role of ACPAs and citrullination in OC activation, and to identify key cellular mediators in this process. Results of our study provide a novel insight into how OC activation might be an initiating event responsible for bone resorption but potentially also for others symptoms related to ACPAs and RA.
Methods
Patients
Detailed demographic characteristics are included in the online supplementary file S1.
ACPA generation
Total IgGs from the synovial fluid (SF, n=25) and peripheral blood (PB, n=35) of patients with RA were isolated on protein G followed by ACPA IgG affinity purification on CCP2 columns as described previously.23 Monoclonal ACPAs RA1103:01:B02 (B02), RA1276:01:D10 (D10), RA 1325:01:B09 (B09) and RA1276:01:C07 (C07), monoclonal RF (RA1276:01:C11) and anti-tetanus toxoid antigen aa1300-1314 control monoclonal antibody RA1362:01:E02 (E02) were isolated from single B-cells isolated from the SF of patients with ACPA-positive RA as previously described.24 Monomeric Fab fragments of B02, D10 and E02 monoclonal antibodies were obtained using the same methodology. The Fc part was exchanged for a murine IgG2a Fc part to generate murinised mE02, mB02, mD10 and mC0724 for use in immunohistochemistry. All of the antibody preparations were endotoxin free.
Cell cultures
Monocytes were isolated from either the blood donor buffy coats or the PB of patients with ACPA-positive RA (n=6) by Ficoll separation (Lymphoprep; Axis Shield, Norway) and selection with anti-CD14 microbeads (Miltenyi Biotec Norden, Lund, Sweden). CD14-positive monocytes were differentiated into Mφ in Dulbecco's modified Eagle medium supplemented with 25 ng/mL macrophage colony-stimulation factor (M-CSF) (Peprotech, London, UK) for 3 days, and further maturated into OCs in the presence of M-CSF (concentration range 10–30 ng/mL) and RANKL (concentrations range 2.5–5 ng/mL; R&D Systems, Abingdon, UK). Half of the medium was replaced every three days. OCs were analysed using tartrate-resistant acid phosphatase (TRAP) staining (leucocyte acid phosphatase kit 387A, Sigma-Aldrich, Stockholm, Sweden). TRAP-positive cells with at least three nuclei were counted as OCs using a light microscope. OCs were grown in parallel on synthetic calcium phosphate-coated plates (Corning, New York, USA). Erosions were visualised under a light microscope and quantified by measuring the resorption area in two random fields per well under low magnification using NIS elements software (Nikon Instruments Europe BV, Amsterdam, the Netherlands) after 14–18 days.
Synovial fibroblasts were isolated from the synovial tissue of patients with RA obtained at the time of joint replacement (n=2). Synovial fibroblasts were grown to 80% confluence on collagen precoated plates and scratches were made, followed by 48 h incubation with or without PAD inhibitor. Images taken immediately at 0 and 5 h after scratching were analysed using NIH ImageJ. The closure areas were normalised to medium controls, and these values represent the migration index.
Pro-inflammatory cytokines/chemokines were analysed using cytometric bead array kits (CBA, BD Biosciences, San Diego, California, USA). IL-8 was neutralised in the cell supernatants using an anti-IL-8/CXCL8 neutralising antibody (clone MAB208, R&D Systems) and PAD activity was inhibited using a pan-PAD inhibitor Cl-amidine (Cayman Chemical, Michigan, USA). The lactate dehydrogenase (LDH) levels in culture supernatants were measured using an LDH cytotoxicity assay kit (Roche Diagnostics Scandinavia AB, Bromma, Sweden). All cell culture media were supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 100 IU/mL penicillin and 50 μg/mL streptomycin (Sigma-Aldrich).
PAD activity assay
Cell pellets were lysed using lysis buffer containing EDTA-free protease inhibitor followed by sonication for 5 min and centrifugation for 15 min. Protein concentrations were measured using a DC protein assay (BIO-RAD, Stockholm, Sweden). PAD activity was measured using an antibody-based assay (ABAP; Modi Quest Research, the Netherlands).25 Cell lysates were added to arginine-coated plates and the deiminated arginine was measured using MQR mouse anti-deiminated arginine antibodies. Colorimetric changes were determined at 450 nm in a multiwell plate reader.
Immunohistochemical analysis and confocal microscopy
Cells were fixed with formaldehyde (Sigma-Aldrich) and stained with murinised monoclonal ACPAs (D10, B02, C07), murinised control antibody (E02), rabbit polyclonal anti-PAD2 (Cosmo Bio, Tokyo, Japan) and monoclonal mouse anti-PADI4 (Abcam, Cambridge, UK) followed by horseradish peroxidase conjugated antimouse antibody as a secondary antibody and 3,3-diaminobenzidene (DAB). The slides were counterstained with Mayer's haematoxylin and viewed using a light microscope (Reichert Polyvar 2 type 302001, Leica). For confocal microscopy (Leica TCS SP5 Microscope), the cells were incubated with the murinised ACPAs, polyclonal ACPA and anti-CD68 monoclonal antibody, followed by Alexa-Fluor-633-labeled secondary antibodies (Abcam) and counterstained with 4′,6-diamidino-2-phenylindole (Sigma-Aldrich).
Animal experiments
Animal experiments were conducted using adult male Balb/c (Harlan) 15 weeks of age. Mice were housed in standard cages (3–5 per cage) in a climate-controlled environment maintaining a 12 h light/dark cycle with access to food and water ad libitum. All experiments were approved by the local ethics committee for animal experiments in Sweden. Mice were injected (intravenously) with either saline or mAb ACPA (2 mg, equal mixture of D10 and B02) diluted in 100 μL saline. Starting day 6, the CXCR1/CXCR2 antagonist reparixin (L-lysin salt, HY-15252, MedChem Express) was injected subcutaneously (in 100 μL saline) twice daily (30 mg/kg/day) for 6 days. At the end of the study, mice were anaesthetised using 4% isoflurane, decapitated and left hind leg removed and post-fixed in 4% paraformaldehyde (PFA) until further analysis. C-terminal telopeptide type 1 collagen was measured in the mice serum using the Ratlaps EIA kit (Immunodiagnostic Systems, UK). Bone structure was blindly analysed using a SkyScan 1176 micro-CT (Bruker) with a voxel size of 9 μm (for detailed protocol, see online supplementary file S1) by two observers (TJ and MM) as previously described.26 ,27
Statistical analysis
Mean differences between groups were compared using either a one-way or two-way analysis of variance followed by Tukey's post hoc test using GraphPad Prism V.6 software. p Values <0.05 were considered significant.
Results
Polyclonal ACPAs promote osteoclastogenesis
Polyclonal ACPAs obtained from either PB or SF reacted with a large number of different cit-peptides from different putative autoantigens as detected by a multiplex chip-based assay28 (see online supplementary figure S2). PB-derived as well as SF-derived affinity-purified ACPA IgG pools, but not control IgGs (flow-through fractions of the CCP-2 affinity columns; ie, CCP-2 non-reactive IgGs), were effective in inducing osteoclastogenesis from PB-derived Mφ of healthy individuals (a mean fold increase in the OC numbers of 1.9±0.3 for PB-derived ACPA and 1.9±0.2 for SF-derived ACPAs compared with those of controls; p<0.05, figure 1B). Similar results were obtained when OCs were obtained from PB-derived Mφ of patients with ACPA-positive RA (data not shown).
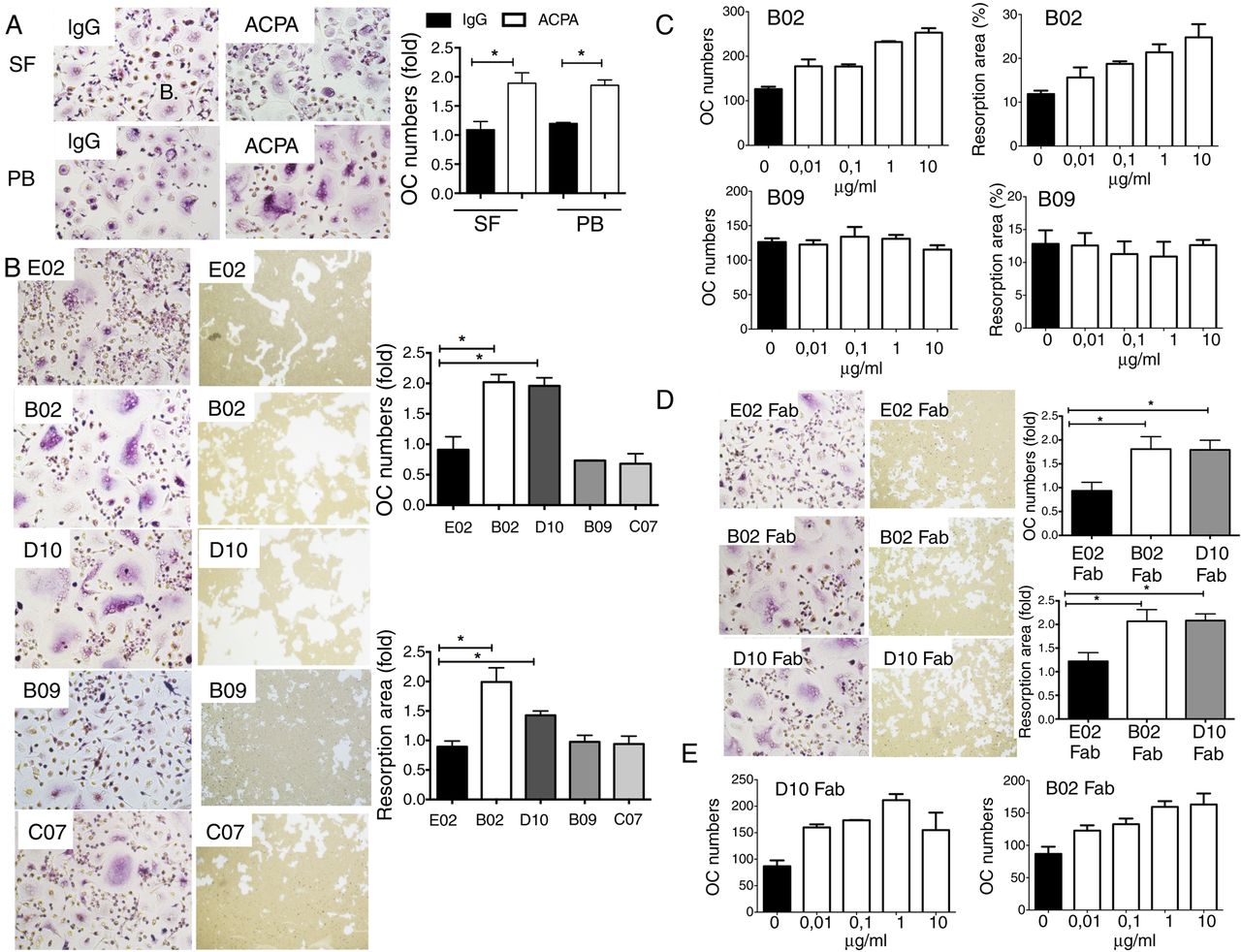
Polyclonal (anti-CCP2 affinity-purified) and monoclonal (single B-cell-derived) anti-citrullinated protein/peptide antibodies (ACPAs) induce osteoclast (OC) activation and bone resorption. (A) Tartrate-resistant acid phosphatase (TRAP) staining of mature OCs obtained from Mφ derived from CD14-positive monocytes of healthy individuals and cultured in the presence of either non-ACPA flow-through IgGs or ACPA IgGs (ACPA) at a concentration of 0.1 µg/mL (original magnification 200×). The graph represents the fold increase in OC (TRAP-positive cells with ≥3 nuclei) numbers and fold increase in resorption areas. The values represent the mean±SEM of three independent experiments. (B) TRAP staining of mature OCs and microscopic visualisation of calcium phosphate resorption areas in the presence of four monoclonal ACPAs (ie, B02, D10, B09 and C07) and one control anti-tetanus monoclonal antibody (ie, E02) at a concentration of 1 µg/mL. The graphs represent fold increases in OC (TRAP-positive cells with ≥3 nuclei) numbers and fold increases in resorption area. The values represent the mean±SEM of four independent experiments. (C) Graphs represent the mean±SEM of dose titrations experiments of stimulatory B02 and non-stimulatory B09 ACPA in OC (number of TRAP-positive cells with ≥3 nuclei) and bone resorption assay (resorption area in %). (D) TRAP staining of mature OCs and microscopic visualisation of calcium phosphate resorption area in the presence of Fab fragments of D10, B02 and E02 antibodies (1 µg/mL). (N=4). The graphs represent fold increases in OC (TRAP-positive cells with ≥3 nuclei) numbers and fold increases in resorption area. The values represent the mean±SEM of four independent experiments. *p<0.05.
Epitope specificity of monoclonal ACPAs derived from single SF B-cells is essential for their capacity to activate OCs
We further tested the effect of individual monoclonal ACPAs derived from single B-cells of ACPA-positive RA SF. The control E02 antibody, two of the ACPA monoclonal antibodies (B09 and C07) and monoclonal RF showed no effect on either osteoclastogenesis or bone destruction (figure 1B, C and online supplementary figure S3). In contrast, two other ACPA monoclonals (D10 and B02) enhanced both OC formation (a fold increase of 2.0±0.1 for both B02 and D10 compared with the control E02 antibody, figure 1B) and the bone resorption area (a fold increase of 2.0±0.2 for B02 and 1.4±0.1 for D10 compared with the control E02 antibody, figure 1B) in a dose-dependent manner (figure 1C). Notably, ACPAs that induced OC activation (B02 and D10) react with the immunodominant cit-epitopes of enolase and vimentin (CEP1 and cit vim 60–75), whereas ACPAs that failed to induce OC activation (B09 and C07) mainly reacted with other cit-peptides, such as those derived from fibrinogen.
We further tested the effect of monomeric Fab fragments showing that Fab fragments of both D10 and B02, but not E02 antibodies, were able to promote osteoclastogenesis (a fold increase of 1.8±0.3 for B02 and 1.8±0.2 for D10, figure 1D) and in vitro bone destruction (a fold increase of 2.1±0.2 for B02 and 2.1±0.1 for D10, figure 1D) in a dose-dependent manner (figure 1E).
PAD enzymes and citrullination are essential for OC differentiation and ACPA-induced activation
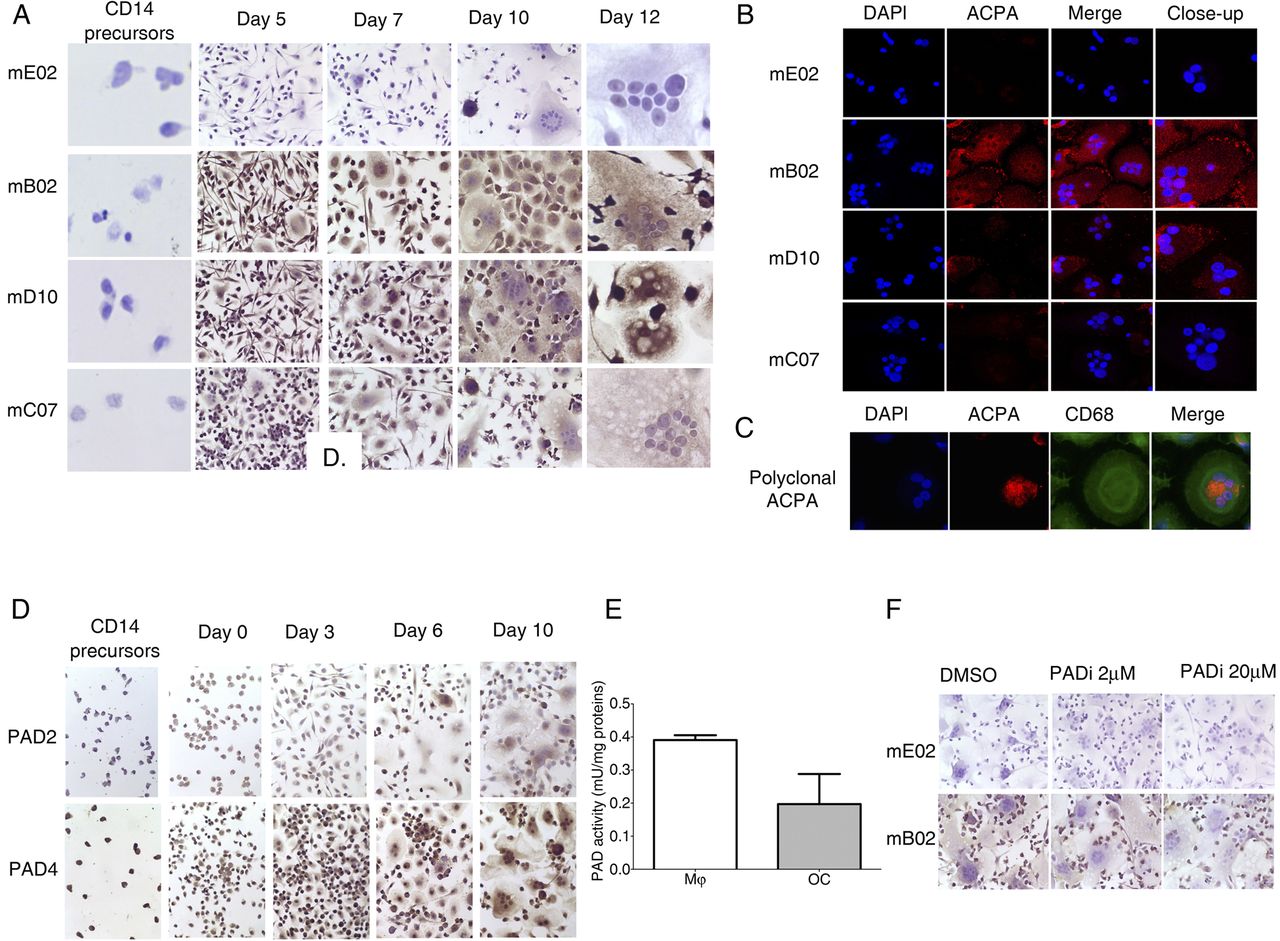
The osteoclastogenic effect of ACPAs but not of the control antibodies suggested that citrullination might be an important event in OC development. We, therefore, investigated citrullination patterns during OC development showing that both Mφ precursors and mature OCs stained positively for the B02 and D10, but neither for C07 ACPA nor for E02 control monoclonal antibodies. No staining with either of the antibodies was detected in the CD14-positive cells from which Mφ were originally developed (figure 2A). Confocal microscopy confirmed biding of B02 and D10 ACPA (figure 2B) as well as polyclonal ACPA (figure 2C) on the OC's cellular surface.

Expression of citrullinated targets and peptidylarginine deiminases (PAD) enzymes during different stages of osteoclast (OC) differentiation. (A) immunohistochemistry images showing brown 3,3-diaminobenzidene (DAB) staining of citrullinated targets in different stages of differentiation from CD14-positive monocyte precursors to Mφ and mature OCs. Slides were stained with murinised monoclonal anti-citrullinated protein/peptide antibodies (ACPAs) (mB02, mD10, mC07) and a monoclonal control antibody (mE02) and counterstained with haematoxylin (original magnification 500× for CD14-positive monocytes and mature OCs and 250× for the intermediate stages). (B) Confocal microscopy images showing red fluorescence staining with monoclonal murinised ACPAs (mE02, mB02, mD10, mC07) and blue 4′,6-diamidino-2-phenylindole (DAPI) nuclear staining in mature OC. (C) Confocal microscopy images showing red fluorescence staining with polyclonal ACPAs, green fluorescence with anti CD68 antibody and blue nuclear staining with DAPI in mature OCs. (D) Immunohistochemistry images showing brown DAB staining of PAD2 and PAD4 expression in different stages of differentiation from CD-14-positive monocyte precursors to Mφ and mature OCs. Slides were counterstained with haematoxylin (original magnification 250×). (E) PAD activity was measured using an antibody-based assay by adding Mφ and OC cell lysates to arginine-coated plates, followed by ELISA measurement of the amounts of deiminated arginine. The graph represents the PAD enzyme activity expressed in mU/mg protein. The values represent the mean±SEM of two independent experiments. (F) Immunohistochemistry images showing brown DAB staining of citrullinated targets in mature OCs with or without incubation with a PAD inhibitor (Cl-amidine) added from the beginning of the cultures. Slides were stained with murinised monoclonal ACPAs (mB02) and a monoclonal control antibody (mE02) and counterstained with haematoxylin (original magnification 250×).
Subsequently, we investigated the expression patterns of PAD enzymes. Immunohistochemistry demonstrated a faint staining in CD14 monocytes with increased staining intensity in both Mφ precursors and more mature OCs for PAD-2 and a more constant expression of PAD4 through different stages of OC maturation (figure 2D). Significant PAD activity was detected during all stages (figure 2E). Addition of the PAD2/PAD4 inhibitor (PADi) Cl-amidine decreased the ACPA binding to the OCs (figure 2F).
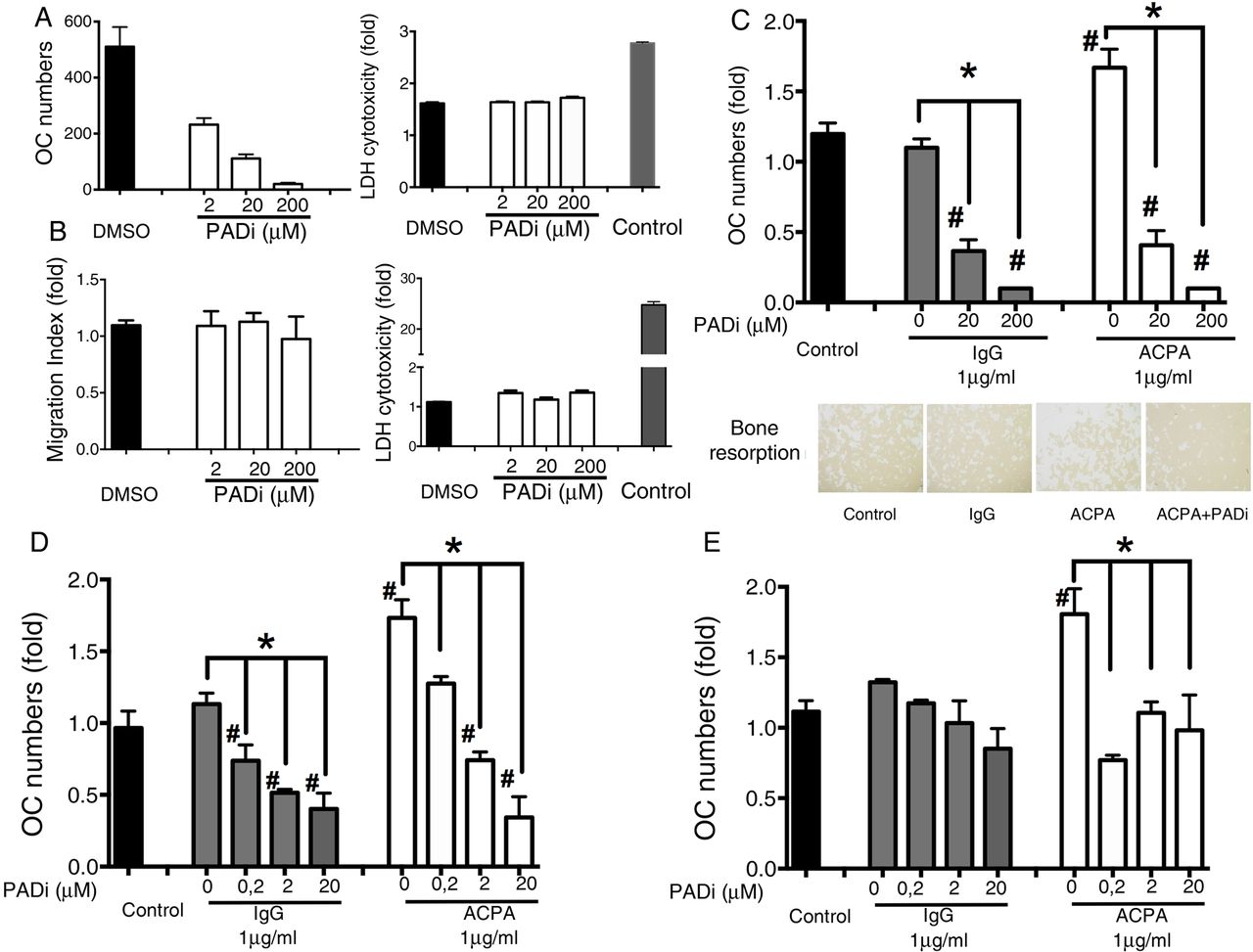
PADi dose-dependently inhibited OC differentiation despite presence of RANKL, without affecting cell viability (figure 3A). In contrast, no changes in cell phenotype (fibroblast migration) or survival were observed when RA-derived synovial fibroblasts (used as a control cell population) were incubated with PADi at similar doses (figure 3B).

Peptidylarginine deiminases (PAD) enzymes are essential for osteoclastogenesis and the anti-citrullinated protein/peptide antibody (ACPA)-mediated effect. (A) PAD inhibition (PADi, Cl-amidine) dose-dependently inhibited osteoclast (OC) differentiation and maturation without any cytotoxic effect. The graphs represent fold decreases in OC (tartrate-resistant acid phosphatase (TRAP)-positive cells with ≥3 nuclei) numbers and fold increases in LDH release in the culture supernatants. The values represent the mean±SEM. (B) PADi does not affect either SF migration or survival. The graphs represent fold increases in the migration index of synovial fibroblast and LDH release in the culture supernatants. The values represent the mean±SEM. (C) the addition of PADi from the beginning of the OC cultures prevented ACPA-induced OC activation and calcium phosphate resorption. The graphs represent fold increases in OC (TRAP-positive cells with ≥3 nuclei) numbers. The values represent the mean±SEM of three independent experiments. Images represent the resorption area by OCs (original magnification 40×). (D) Dose titration of PADi showing that early PAD inhibition (at the initiation of the OC culture) with doses as low as 0.2 μM PADi inhibits ACPA-mediated osteoclastogenesis but no longer the unstimulated differentiation of OCs. The graphs represent fold decreases in OC (TRAP-positive cells with ≥3 nuclei) numbers. The values represent the mean±SEM. (E) Late PAD inhibition (3 days before ending the OC cultures) inhibited ACPA-mediated osteoclastogenesis but not the unstimulated differentiation of OCs. The graphs represent fold increases in OC (TRAP-positive cells with ≥3 nuclei) numbers. The values represent the mean±SEM. *p<0.05.
PADi addition at the beginning of OC cultures completely prevented OC activation and bone resorption with or without ACPAs (figure 3C). Further dose titration experiments showed, however, that doses as low as 0.2 μM PADi only inhibit ACPA-mediated OC activation, but not the differentiation of OCs without ACPAs (figure 3D). Time kinetic experiments showed that early PADi addition (at the initiation of the OC culture) inhibited OC activation with and without ACPAs (figure 3D), while late PADi inhibition (3 days before ending the OC cultures) inhibited only ACPA-mediated OC activation (figure 3E).
IL-8 is an essential mediator of ACPA-driven OC activation
To investigate potential mediators responsible for the effect of ACPAs, we analysed a set of common cytokines in cell culture supernatants. IL-6, IL-1, IL-10 and tumour necrosis factor (TNF)-α were detected at low basal levels and showed no consistent changes during OC development with or without ACPA treatment (data not shown). In contrast, IL-8 levels were significant increase in the OC supernatants of ACPA-treated OCs (figure 4A and online supplementary figure S4). Time titration experiments revealed that high levels of IL-8 were detected in Mφ-derived OC cultures at early time points during their maturation (2426±29 pg/mL at day 4) and further increased with time (5532±98 pg/mL at day 6 and 9858±387 pg/mL at day 12). ACPAs, but not control IgGs, further increased IL-8 release in the culture supernatants over time (figure 4B). Added IL-8 in the absence of increased osteoclastogenesis (figure 4C). Blockade of extracellular IL-8 with a neutralising IL-8-specific antibody in the presence of M-CSF and RANKL dose-dependently blocked the differentiation of immature into mature OCs (figure 4D) and was also able to block the effects of ACPA at doses as low as 1 μg/mL (figure 4E). ACPAs’ effects were blocked when the neutralising anti-IL-8 antibodies were added either at the beginning (the first three days) or at the end of the cultures (the last three days) (figure 4F). No such effects were observed when TNF-α was blocked with adalimumab, even at higher concentrations (10 μg/mL, figure 4G).

Interleukin (IL)-8 is an essential mediator of anti-citrullinated protein/peptide antibody (ACPA)-driven osteoclastogenesis. (A) cytometric bead array showed that ACPA, but not control IgGs, increased IL-8 release in the culture supernatants of mature osteoclasts (OCs). The values represent the mean±SEM of three independent experiments. (B) Cytometric bead array showed high levels of IL-8 in Mφ-derived OC cultures at early time points during their maturation, which further increased over time. ACPA, but not control IgGs, additionally increased IL-8 release in the culture supernatants at all time points tested. The graph shows a representative time kinetic variation in IL-8 concentrations in cell culture supernatants from one of the three tested donors. The values represent the mean±SEM. (C) Exogenous added IL-8 increases osteoclastogenesis in a dose-dependent manner. The values represent the mean±SEM. (D) Neutralising anti-IL-8 antibodies inhibited Mφ-derived OCs maturation dose dependently. The graphs represent fold decreases in OC (tartrate-resistant acid phosphatase (TRAP)-positive cells with ≥3 nuclei) numbers. The values represent the mean±SEM of three independent experiments. (E) Anti-IL-8 neutralising antibodies completely abolished the effect of ACPAs at doses as low as 1 μg/mL. The graphs represent fold increases in OC (TRAP-positive cells with ≥3 nuclei) numbers. The values represent the mean±SEM of three independent experiments. (F) Both early (first three days of culture) and late (last three days of the culture) addition of anti-IL-8 neutralising antibodies (1 μg/mL) completely abolished the effect of ACPAs. The graphs represent fold increases in OC (TRAP-positive cells with ≥3 nuclei) numbers. The values represent the mean±SE. (G) Anti-IL-8 neutralising antibodies but not an antibody against tumour necrosis factor (TNF)-α (adalimumab) abolished the effect of ACPAs at concentrations as high as 10 μg/mL. The graphs represent fold increases in OC (TRAP-positive cells with ≥3 nuclei) numbers. The values represent the mean±SEM of three independent experiments. *p<0.05.
In vivo ACPA-induced systemic bone loss is reversed by an IL-8 antagonist
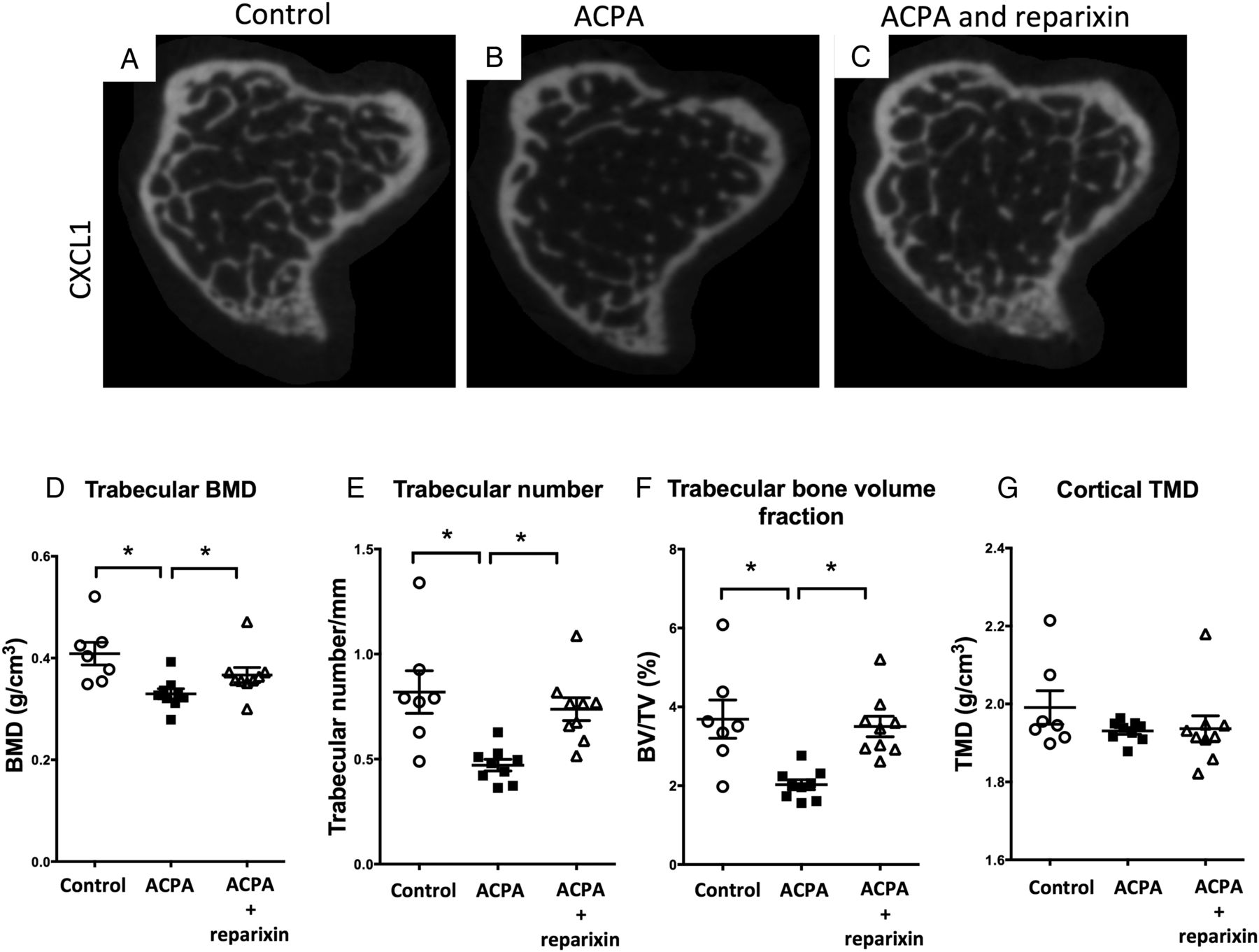
We next tested whether ACPAs can induce bone loss in vivo using micro-CT evaluation of the tibia in control mice (figure 5A) and mice injected with murinised monoclonal ACPAs alone (figure 5B) or together with a CXCR1/2 antagonist (reparixin) blocking the murine IL-8 homologues (figure 5C). ACPA intravenous injection significantly decreased the trabecular bone mineral density (figure 5D), the trabecular number (figure 5E) and the bone volume fraction (bone volume/tissue volume, figure 5F), while not affecting the cortical tissue mineral density (figure 5G). Changes were reversed by subcutaneous administration of reparixin (figure 5D–F). No significant changes were observed in the levels of serum bone catabolism markers or pro-inflammatory cytokines (data not shown). Histological examination of joint tissues revealed minimal signs of synovitis (synovial inflammatory infiltration) and erosions in only one of the 9 ACPA-treated mice, whereas no changes were seen in joint tissues from the other 16 animals.

Anti-citrullinated protein/peptide antibodies (ACPAs) induce systemic bone loss in vivo that is reversed by interleukin (IL)-8 inhibition. Representative 2D micro-CT images of the tibial metaphysis of control mice (A, n=7) and mice that were injected with ACPAs in the absence (B, n=9) or presence of reparixin (C, n=9). (B) Graphs showing quantitative evaluation of the trabecular bone mineral density (BMD, D), trabecular number (E), bone volume fraction (bone volume/tissue volume, F) and the cortical tissue mineral density (TMD, G). The values represent the mean±SEM. *p<0.05.
Discussion
We provide evidence that IL-8 is a key mediator of ACPA-induced OC activation. We show that IL-8 release is prominent after OCs stimulation by ACPAs, and that blockade of IL-8 or its receptor(s) completely inhibits this effect, preventing bone loss both in vitro and in vivo. Furthermore, PAD enzymes are essential for both OC differentiation and ACPA-induced OC activation. Taken together our findings provide novel insights into how OCs might act as first targets for ACPAs and suggest that IL-8 and/or PAD enzymes targeting may be beneficial in very early stages of development of ACPA-positive arthritis.
We show that both serum and joint-derived ACPAs, but not other IgGs, have the capacity to activate OCs and that epitope specificity is important for this effect. The substantial cross-reactivity of the SF single B-cell-derived monoclonal ACPAs for different cit-epitopes previously described24 prevents us from more detailed characterisation of the epitopes that are crucial for OC activation. The OC-inducing capacity of Fab fragments adds to previous data on the importance of Fc configuration for OC activation,29 showing that both epitope specificity and Fc structure has to be taken into account in identifying ACPAs with various effects (activating, neutral or inhibitory). The mechanisms involved in ACPA-induced OC activation have so far been relatively unknown. OCs can produce many different cytokines/chemokines after exposure to pro-inflammatory stimuli.30 IL-8 production by OCs has been described previously,31 and it was recently proposed that IL-8 has an autocrine effect on osteoclastogenesis20 but not in the context of ACPA stimulation. Our findings show that IL-8 is the dominating cytokine/chemokine (out of the standard set measured here) released from ACPA-stimulated OCs, adding a new dimension to these earlier findings. These new data show that exposure of OCs to ACPAs results in the preferential release of IL-8, but not other common pro-inflammatory cytokines, indicating that ACPAs specifically induce production of IL-8 from OCs and that IL-8 mediates an autocrine activation of these same cells.
The gradually increased OC expression of both cit-epitopes as well as PAD2 and PAD4 enzymes, and the dose-dependent OC inhibitory effect of PADi, even in the absence of ACPAs indicate that PADs, and thus citrullination, have unique functions during OC differentiation that are not present in other cells (as shown here for synovial fibroblasts). Thus, presence of cit-epitopes within and on the cell surface of OCs during their normal differentiation, in contrast to other cells that express cit-proteins mainly in the context of inflammation,16 might, therefore, explain how OCs can be preferentially targeted by ACPAs in a non-inflammatory context.
There are some caveats to the current study. For example, the detailed signalling pathways contributing to OC activation by ACPAs, as well as the targets of the specific ACPAs, remain to be identified. Further, the potential synergy between ACPA, IL-8 and others inflammatory stimuli that might contribute to the transition from bone loss to long-lasting joint inflammation needs further investigation. Even though bone damage has been associated with ACPA before RA onset32 and could be present in patients with clinically inactive treated RA,21 the complex interaction between synovial inflammation and bone loss still needs to be addressed.
In conclusion, our observations enable us to propose a novel, testable hypothesis for how ACPAs might specifically target the joints (figure 6). Thus, the cell-specific requirement of PAD for OC differentiation leads to local expression of cit-epitopes, allowing specific targeting of OC precursors by circulating ACPAs. This leads to increased amounts of IL-8 that further stimulate OCs through an autocrine loop, resulting in a first step in bone loss and also pain (as shown in Wigerblad et al, submitted simultaneously). The communication system between bone marrow and synovium33 allows IL-8 to migrate to the joint. One possibility that still remains to be demonstrated is that these events contribute to secondary chemoattraction and activation of other inflammatory cells eventually including neutrophils, which might be further activated by ACPAs to release neutrophil extracellular traps.34 Such a scenario where OCs are the primary target of ACPAs and upon stimulation become able to initiate a local inflammatory cascade might help answer the long-standing questions regarding why and how ACPAs may specifically contribute to joint inflammation and not inflammation elsewhere, and why initial lesions often occur at the site where bone and synovium meet. Interestingly, our results also open the way for preclinical studies to test the therapeutic and preventive effect of PADi and IL-8-blocking agents in models of RA and eventually also in ACPA-positive individuals at risk of developing RA.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic illustration of the peptidylarginine deiminases (PAD)-dependent differentiation and maturation of osteoclasts (OCs), allowing initial OC targeting by anti-citrullinated protein/peptide antibodies (ACPAs) with consecutive interleukin (IL)-8 release. OC precursors (OCPs) are present in the bone marrow and can develop into mature OCs. During the differentiation and activation of OCP, a gradual increase in cell citrullination occurred as a consequence of increased PAD activity in a calcium-rich microenvironment. ACPAs present in the circulation can reach and bind to maturing OCPs in the bone marrow, leading to an increase in OC activity with consecutive bone resorption through an IL-8-dependent autocrine loop. In a second step, IL-8 will reach the joint and initiate the chemoattraction and migration of inflammatory cells in particular neutrophils. Neutrophil extracellular traps are released by these neutrophils in the presence of ACPAs, which further contributes to the initiation of joint inflammation with the local accumulation of other inflammatory cells (such as macrophages) and activation of synovial fibroblasts, resulting in synovial membrane inflammation. NET, neutrophil extracellular traps.
Acknowledgments
The authors acknowledge the excellent technical assistance from Hana Hailu, Emily Barry, Lena Israelsson and Monika Hansson.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
- Data supplement 1 - Online supplement
- Data supplement 2 - Online supplement
Footnotes
Handling editor Tore K Kvien
Correction notice This article has been corrected since it published Online First and in print. The conclusion section of the abstract has been amended.
Contributors AK and AIC designed the experiments, analysed the data and wrote the manuscript along with input from VM, HW, TJ, CS, AHH, S-BC and LK. AK conducted all osteoclast assays with help from VJ and NP-V. AK, VJ and HW measured chemokines and cytokines in supernatants. TJ, MM, GW, JK and CS performed animal experiments. MS and S-BC designed and performed the fibroblast experiments. ME and AK performed all immunohistochemistry experiments. AHH recruited patients and characterised all clinical data. CF-C, KL, P-JJ and HW purified polyclonal ACPAs. SR, KT, KA and VM produced monoclonal ACPAs. AIC, AK, VJ, HW, CS, S-BC, P-JJ, AJY, VM and LK discussed and developed the concept. All authors critically reviewed and approved the final form of the manuscript.
Funding This work was supported by the Swedish Research Council, FP7-HEALTH-2012 INNOVATION-1 Euro-TEAM (305549-2), the Initial Training Networks 7th framework programme Osteoimmune (289150), and Innovative Medicine Initiative BTCure (115142-2) and through the Regional Agreement on Medical Training and Clinical Research (ALF) between Stockholm County Council and Karolinska Institutet.
Competing interests None declared.
Patient consent Obtained.
Ethics approval Ethical Review Committee of Karolinska University Hospital.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Editorial
- Basic and translational research
- Correction