Article Text

Abstract
Objectives The efficacy and safety of sifalimumab were assessed in a phase IIb, randomised, double-blind, placebo-controlled study (NCT01283139) of adults with moderate to severe active systemic lupus erythematosus (SLE).
Methods 431 patients were randomised and received monthly intravenous sifalimumab (200 mg, 600 mg or 1200 mg) or placebo in addition to standard-of-care medications. Patients were stratified by disease activity, interferon gene-signature test (high vs low based on the expression of four genes) and geographical region. The primary efficacy end point was the percentage of patients achieving an SLE responder index response at week 52.
Results Compared with placebo, a greater percentage of patients who received sifalimumab (all dosages) met the primary end point (placebo: 45.4%; 200 mg: 58.3%; 600 mg: 56.5%; 1200 mg 59.8%). Other improvements were seen in Cutaneous Lupus Erythematosus Disease Area and Severity Index score (200 mg and 1200 mg monthly), Physician's Global Assessment (600 mg and 1200 mg monthly), British Isles Lupus Assessment Group-based Composite Lupus Assessment (1200 mg monthly), 4-point reductions in the SLE Disease Activity Index−2000 score and reductions in counts of swollen joints and tender joints. Serious adverse events occurred in 17.6% of patients on placebo and 18.3% of patients on sifalimumab. Herpes zoster infections were more frequent with sifalimumab treatment.
Conclusions Sifalimumab is a promising treatment for adults with SLE. Improvement was consistent across various clinical end points, including global and organ-specific measures of disease activity.
Trial registration number NCT01283139; Results.
- Systemic Lupus Erythematosus
- Treatment
- Autoimmune Diseases
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Introduction
Systemic lupus erythematosus (SLE) is a chronic, multisystem, autoimmune disease that predominantly affects women of childbearing age. Its clinical manifestations (eg, arthritis, rashes, alopecia, vasculitis, nephritis, serositis) lead to significant morbidity, reduced physical function, loss of employment, impaired quality of life, high risk of permanent disability and shortened life span.1–3 Treatment of SLE remains challenging because of the suboptimal efficacy of standard-of-care medications and the serious adverse events associated with their use.4–6
Several studies, reviewed elsewhere,7–9 have suggested a role for interferon-α (IFN-α) in the pathogenesis of SLE.10–16 Distinct patterns of gene expression induced by type I IFN11 ,17–19 are substantially upregulated in blood, skin biopsies and synovial biopsies of patients with SLE compared with healthy controls.20 This study aimed to investigate whether blocking the type I IFN pathway is an effective approach for the treatment of SLE.
Sifalimumab is a fully human, immunoglobulin G1 κ monoclonal antibody that binds to and neutralises the majority of IFN-α subtypes. Clinical trials of sifalimumab have established its safety profile and its suppression of IFN-α-induced genes, and have suggested favourable effects on clinical outcome measures.21 ,22 This phase IIb trial was conducted to evaluate the efficacy and safety of three fixed intravenous dosages of sifalimumab in adults with moderate to severe active SLE with inadequate responses to standard-of-care treatments.
Methods
Eligible patients were male or female, aged 18–75 years, weighing ≥40 kg, fulfilling ≥4 of the 11 American College of Rheumatology (ACR) SLE classification criteria,23 ,24 and receiving stable dosages of one or more of the following: oral prednisone (≤20 mg/day, or equivalent); azathioprine (≤150 mg/day); antimalarial treatment; mycophenolate mofetil/mycophenolic acid (≤3.0 g/day); or subcutaneous/oral methotrexate (≤20 mg/week). Administration of stable dosages of non-steroidal anti-inflammatory drugs was permitted. Prior to enrolment, biological therapies had to be discontinued for a sufficient period to ensure they would no longer have any pharmacodynamic and/or clinical effect.
At screening, patients had to have positive serology for antinuclear (≥1:40), anti-Smith or anti-double-stranded DNA (dsDNA) (≥100 IU/mL) antibodies by the AtheNA Multi-Lyte ANA-II Plus test system (Alere, Waltham, Massachusetts, USA). Patients also had to meet the following disease activity criteria: SLE Disease Activity Index 2000 (SLEDAI-2K)25 score ≥6 with at least two points from a clinical component (excluding SLE headache or organic brain syndrome), a British Isles Lupus Assessment Group (BILAG)-200426 score of ≥1A or ≥2B organ system scores27 and a Physician's Global Assessment (PGA) of disease activity ≥1 (scale: 0 (none) to 3 (severe)). Patients with active and severe lupus nephritis or neuropsychiatric SLE were excluded from the study. At randomisation, the overall SLEDAI-2K clinical component score was required to be at or above the screening value. Additional study exclusion criteria are provided in the online supplementary material.
Supplemental material
Study design
This study (NCT01283139), conducted at 107 sites in 20 countries, consisted of a 52-week, randomised, double-blind, placebo-controlled, parallel-group treatment phase, followed by a 22-week safety follow-up phase, and was conducted between March 2011 and April 2014. All sites received ethics committee or independent institutional review board approval before commencement of the study.
Dosages of oral corticosteroids were required to remain stable throughout the study, with the exception of limited, protocol-defined, oral corticosteroid burst (followed by a taper) for increased SLE disease activity or protocol-permitted oral corticosteroid tapering (see online supplementary material).
The study was monitored by an independent data safety and monitoring board, which included a rheumatologist and an infectious disease expert. An independent external adjudication group confirmed SLE organ system involvement and disease activity at screening, approved randomisation, and monitored assessments and adherence throughout the trial.
This study was conducted in accordance with the principles of the Declaration of Helsinki and the International Conference on Harmonisation Guidance for Good Clinical Practice. Independent ethics committee approval was obtained and all patients provided written informed consent in accordance with local requirements.
Randomisation and masking
Patients were randomised by an interactive voice and web management system (see online supplementary material) 1:1:1:1 to receive intravenous placebo or sifalimumab 200 mg , 600 mg or 1200 mg on days 1, 15 and 29, and every 28 days thereafter. Randomisation was stratified by IFN gene signature at screening, SLEDAI-2K score (<10 or ≥10) at screening, and prespecified geographical regions. Results of an IFN gene-signature test were designated as high or low based on the expression of four IFN-regulated genes (IFI27, IFI44, IFI44L, RSAD2).28 Geographical regions were based on a previous study22 and clinical experience, which demonstrated greater standard-of-care response rates for patients in central and South America, eastern Europe and Asia (region 1), compared with patients in North America, western Europe and South Africa (region 2).
Efficacy and safety evaluations
The primary efficacy end point was the percentage of patients who achieved an SLE Responder Index (SRI(4)) response at week 52. This was defined as: a 4-point improvement in SLEDAI-2K; no clinically significant worsening (≥0.3) in PGA; and no new BILAG-2004 ‘A’ (severe) or >1 new ‘B’ (moderate) organ system scores.29
Secondary efficacy measures, assessed at week 52, included percentage of patients with decreased oral prednisone dosages (≥10 mg/day, or equivalent, at baseline to ≤7.5 mg/day by week 52); ≥4-point reduction in Cutaneous Lupus Erythematosus Disease Area and Severity Index (CLASI)30 ,31 for patients with at least moderate skin involvement (CLASI ≥10); and >3-point improvement in Functional Assessment of Chronic Illness Therapy-Fatigue.32 Other prespecified efficacy end points included SLEDAI-2K25; modified SRI (mSRI) requiring SLEDAI-2K reductions of 5–8 points; BILAG-based Combined Lupus Assessment (BICLA)33; PGA; numbers of swollen and tender joints; dsDNA; and C3 and C4 complement concentrations.
Safety end points included reporting of adverse events (Medical Dictionary for Regulatory Activities, V.17), laboratory assessments and vital signs.
Statistical analysis
Analysis of the primary end point compared response rates at week 52 between each sifalimumab group and placebo using a logistic regression model with independent variables of treatment group and randomisation stratification factors. Patients who withdrew from treatment had increased use of corticosteroids beyond that permitted (see online supplementary material), or initiated or increased immunosuppressant dosage any time after baseline were considered non-responders. Analyses were performed in the modified intention-to-treat (mITT) population (all randomised patients who received any investigational product and had a baseline primary efficacy measurement) and an mITT subpopulation of patients with a high IFN gene signature. The study result was considered positive if the primary end point was met in either of the two study populations.
The type-I error rate (α level) was controlled at approximately 0.10 (two-sided), within each of the populations for the primary end point analysis, by performing a Cochran−Armitage trend test of all treatment groups prior to performing pairwise comparisons between each sifalimumab group and placebo. No multiplicity adjustment for the two study populations or other end points was applied.
One interim analysis was performed when 46% of primary end point information was available. The two-sided α levels of 0.008 and 0.098 were allocated for the interim and final analyses, respectively, using the O'Brien−Fleming-type Lan−DeMets α spending function approach. Thus, a p value of ≤0.098 would be considered statistically significant for the final analysis.
Exploratory post hoc analyses improved in number of affected joints in a subset of patients with severe joint involvement at baseline and assessment of clinical SLEDAI scores. These were analysed using the aforementioned logistic regression model.
The target sample size of 400 (100 per group) was based on providing 88% power at the 0.10 α level to detect at least 20% absolute improvement in SRI(4) response rate at week 52 for sifalimumab relative to placebo, assuming a 40% placebo response rate.
The two-sided α level of 0.1 represents a 5% chance of declaring a positive study when there is no treatment effect (risk of proceeding with an ineffective drug). The power of 88% represents a 12% chance of declaring a negative study when there is a positive treatment effect (risk of discontinuing development of a potentially efficacious drug). This combination of statistical risks was chosen to balance the continuation and discontinuation risks while maintaining a feasible phase IIb study.
All data analyses were conducted using the SAS System (SAS Institute, Cary, North Carolina, USA).
Results
In total, 834 patients were screened, with 432 randomised to treatment (108 placebo, 108 sifalimumab 200 mg, 109 sifalimumab 600 mg, 107 sifalimumab 1200 mg, all monthly). One patient in the 600 mg group had an entry criteria violation and did not receive study treatment. Patient disposition is presented in online supplementary figure S1. Demographics and baseline disease characteristics are presented in table 1.
Demographics and baseline clinical characteristics (mITT population)
Baseline characteristics were similar between groups, with the following exceptions: disease duration was shorter for placebo than for sifalimumab groups (90.4 vs 98.6–103.9 months) and fewer patients in the 600 mg group were receiving mycophenolate (4.6% vs 10.2–12.0%). Baseline SLEDAI−2K and BILAG−2004 scores were similar between the IFN-high and IFN-low populations, and between patients from different regions. However, there were differences between geographical regions in several demographic and clinical characteristics (see online supplementary table S1). Overall baseline disease activity measures were consistent with moderate to severe active SLE.
No neutralising antibodies to sifalimumab were found in any patient over the course of the study.
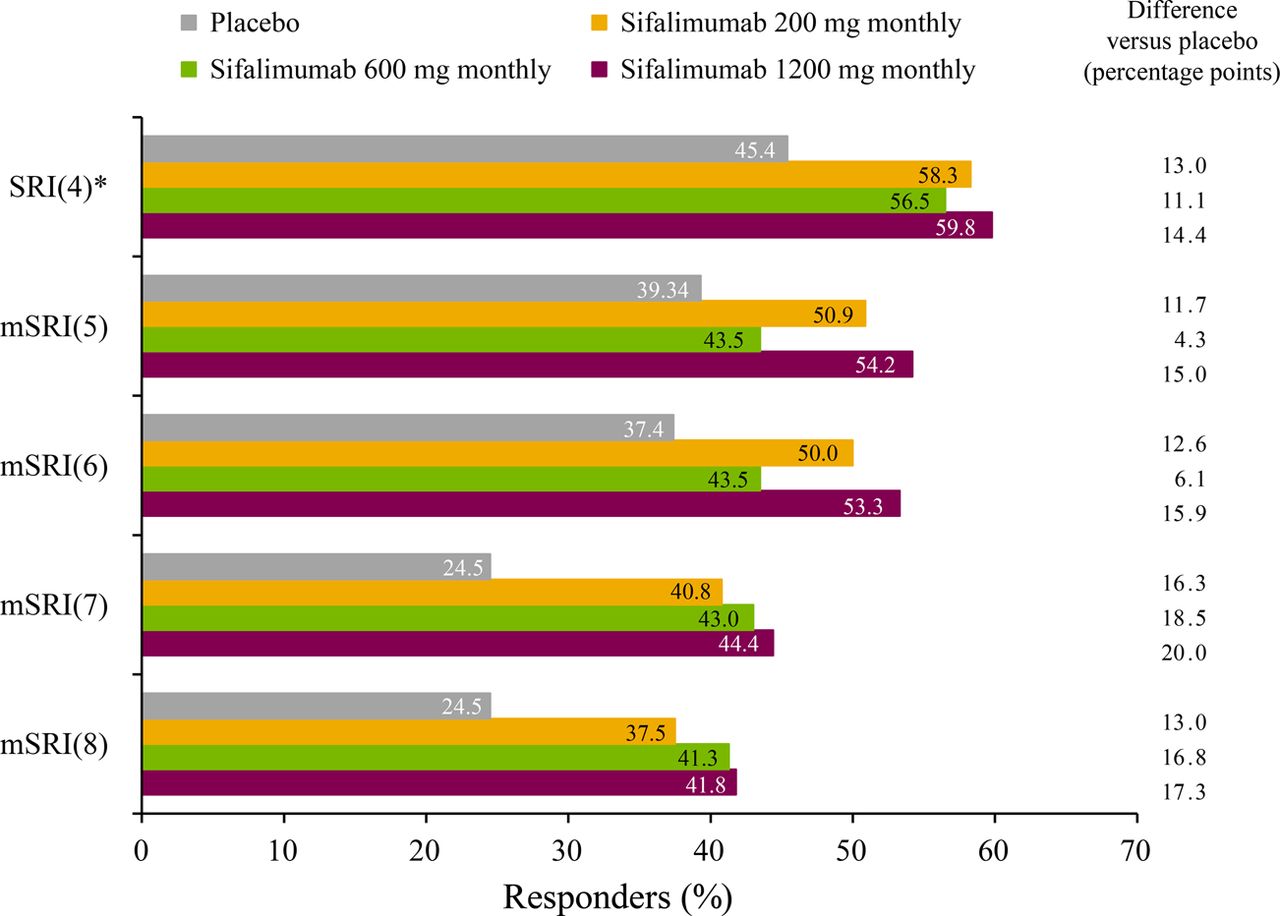
At week 52, the Cochran–Armitage trend test of all treatment groups showed that the number of patients achieving the primary end point was greater for sifalimumab versus placebo (p=0.053). Pairwise comparisons demonstrated that this effect was consistent for each sifalimumab dosage (200 mg monthly: 58.3%, p=0.057; 600 mg monthly: 56.5%, p=0.094; 1200 mg monthly: 59.8%, p=0.031) compared with placebo (45.4%) (table 2) with improvements reaching a peak at week 24, after which there was a plateau in the effect (figure 1). More patients receiving sifalimumab versus placebo also achieved the predefined exploratory mSRI end points (table 2, figure 2).
Percentage of patients achieving efficacy end points at week 52 (mITT population)
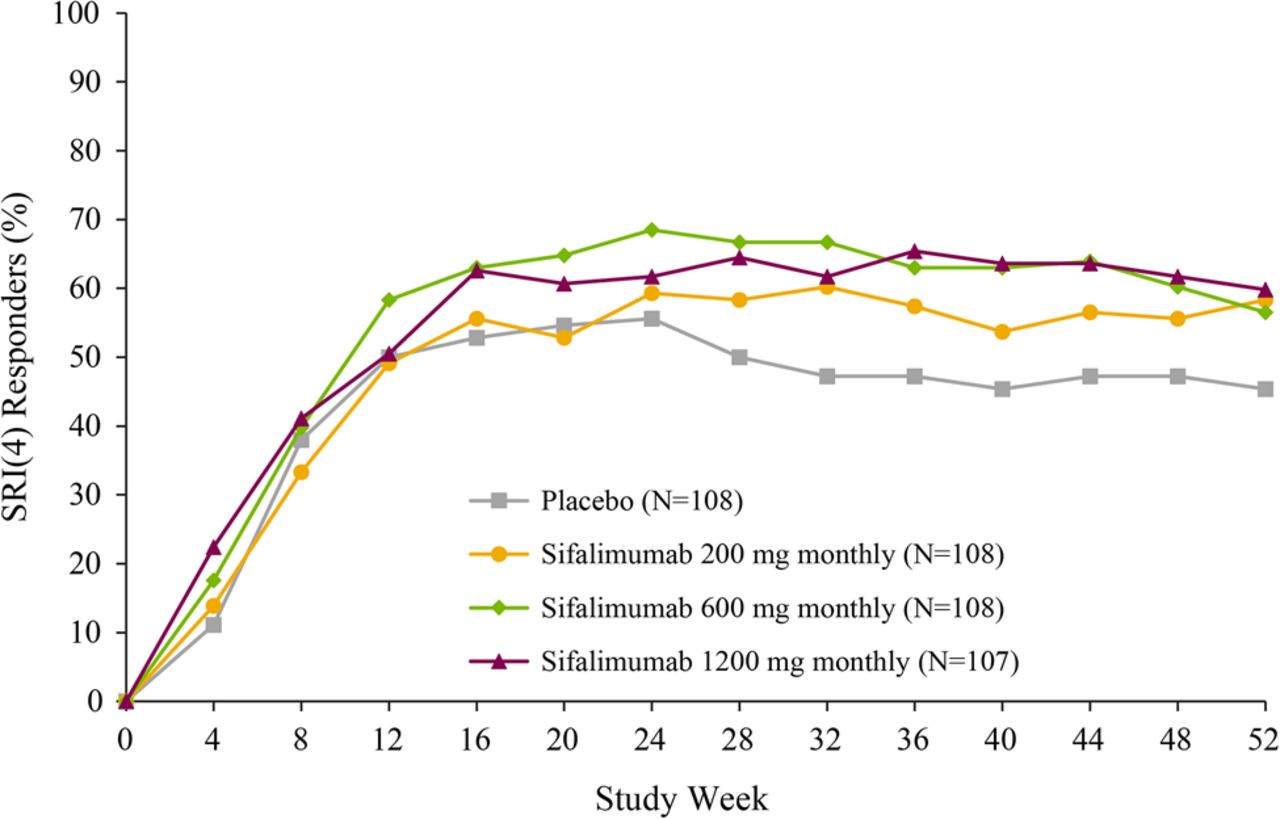
Primary end point: patients achieving a SRI(4) at week 52 (mITT population). Treatment was administered on days 1, 15 and 29, and then every 28 days thereafter. mITT, modified intention-to-treat; SRI(4), systemic lupus erythematosus responder index.

Patients achieving an mSRI response with 5-, 6-, 7-, or 8-point decreases in SLEDAI-2K scores (mITT population). *Primary end point. Treatment was administered on days 1, 15 and 29, and then every 28 days thereafter. mITT, modified intention-to-treat; mSRI, modified systemic lupus erythematosus responder index; SLEDAI-2K, Systemic Lupus Erythematosus Disease Activity Index 2000.
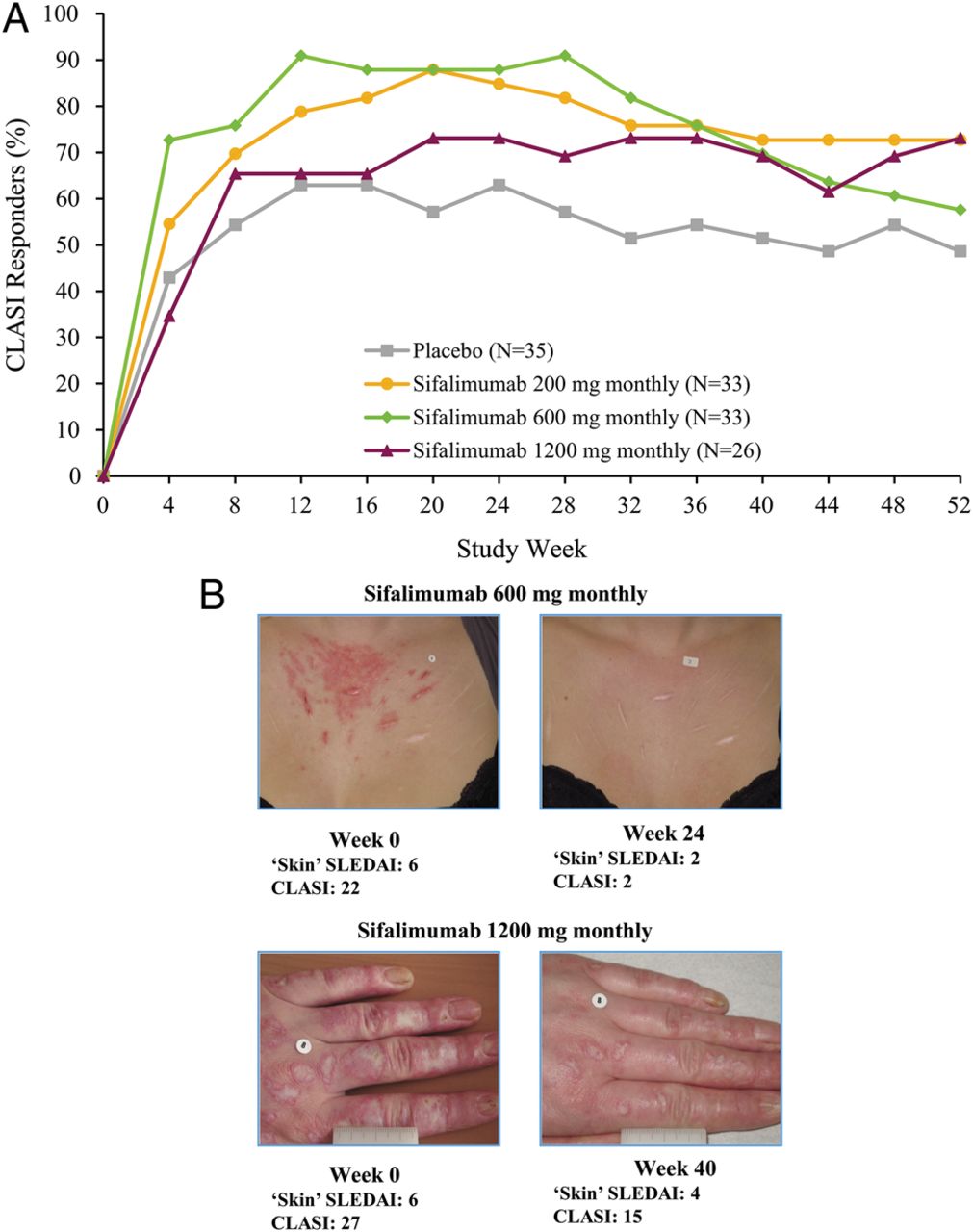
The percentage of patients with improvements in CLASI was greater for all sifalimumab dosages compared with placebo (table 2); maximum improvement seen after 20–24 weeks (figure 3). The percentage of patients receiving oral corticosteroids ≥10 mg/day at baseline who were tapered to ≤7.5 mg/day was low in all groups (6–9%), although substantially more patients met predefined criteria for tapering in the sifalimumab 600 mg and 1200 mg monthly groups compared with placebo (table 2).

{kind=link}
{kind=link}
{kind=link}
Secondary end point. Panel A shows CLASI responder rate (patients with a CLASI activity score greater ≥10 at baseline who achieved a ≥4-point reduction) (mITT population). Panel B shows examples from the 600 mg and 1200 mg groups of skin response following sifalimumab treatment. (Full permission for use of these images has been granted by patients.) Treatment was administered on days 1, 15 and 29, and then every 28 days thereafter. CLASI, Cutaneous Lupus Erythematosus Disease Area and Severity Index; mITT, modified intention-to-treat; SLEDAI, Systemic Lupus Erythematosus Disease Activity Index.
Of the predefined exploratory assessments, all sifalimumab dosages resulted in greater percentages of patients achieving a ≥4-point reduction in SLEDAI-2K and having a positive BICLA response compared with placebo. Trends towards greater improvements with sifalimumab were noted for both the more frequently (mucocutaneous, musculoskeletal) and less frequently (haematological, renal, vascular) involved SLEDAI organ systems (see online supplementary figure S2). Improvements in PGA were also greater for the sifalimumab groups. Rates of disease flares, defined as increased disease activity (new BILAG-2004 ‘A’ or ‘B’ organ system score; SLEDAI-2K score >3; or adverse events reflecting increased SLE disease activity) resulting in an increase in oral corticosteroid dosages, were lower for the 600 mg and 1200 mg monthly sifalimumab groups compared with placebo. No specific differences between the groups in changes from baseline or normalisation of C3/C4 complement concentrations (see online supplementary figure S3) or anti-dsDNA concentrations (see online supplementary figure S4) were observed. The percentage of patients achieving the SRI(4) end point was not influenced by the type of anti-dsDNA assay used (see online supplementary table S2).
Post hoc exploratory analyses demonstrated that in a subset of patients with severe joint involvement (≥eight swollen and ≥eight tender joints) at baseline, there was a dosage-related increase in the percentage of patients with ≥50% decrease in affected joints, which was substantially greater for all dosages versus placebo (table 2). There was also an apparent dosage-related increase in the percentage of patients with clinically meaningful reductions in clinical SLEDAI response with substantial improvements over placebo seen for the 600 mg and 1200 mg monthly dosages.
Subgroup analyses of SRI(4), mSRI(8) and BICLA by IFN test status indicated that substantial improvements were observed for IFN-high patients versus placebo (table 2). However, the small number of IFN-low patients, comprising approximately 19% of the study population, prevented a meaningful statistical comparison between patients based on IFN test status.
Differences were also observed in SRI(4) results in subgroup analyses by geographical region, with higher response rates in region 1 compared with region 2, although a greater distinction between sifalimumab and placebo groups was seen in region 2 (see online supplementary figure S5). No differences between the regions were seen in baseline SLEDAI-2K, PGA or BILAG-2004 scores (data not shown) but, as previously mentioned, there were several differences in baseline demographic and clinical characteristics (see online supplementary table S1).
The percentages of patients with at least one adverse event, serious adverse event or adverse event leading to discontinuation were similar across the groups (table 3). The most common adverse events were worsening of SLE, urinary tract infections, headaches, upper respiratory tract infections and nasopharyngitis. There were five deaths during the double-blind period: two in the placebo group (both septic shock); one in the 600 mg monthly group (cardiopulmonary arrest/sepsis) and two in the 1200 mg group (stab wound, cardiopulmonary arrest/transient ischaemic attack). There was one additional death during the follow-up period (600 mg group; mitral valve failure).
Adverse events (safety population)
One patient (sifalimumab 1200 mg monthly) was diagnosed with active tuberculosis and responded well to treatment. There was one case of progressive multifocal leucoencephalopathy (placebo) and one of viral encephalitis (sifalimumab 600 mg monthly). Two grade 4 (life-threatening) infections were reported with sifalimumab: bacterial enterocolitis (200 mg monthly) and bacterial meningitis (1200 mg monthly). Both resolved without sequelae following treatment. Serious infections were reported in eight (7.4%), nine (8.3%), seven (6.5%) and eight (7.5%) patients on placebo, sifalimumab 200 mg, 600 mg and 1200 mg, respectively. There were no cases of systemic fungal infection.
Herpes zoster infections were reported in 1 (0.9%) patient on placebo compared with 19 (5.9%) patients treated with sifalimumab, with the highest incidence in the sifalimumab 1200 mg monthly group (table 3). All patients responded promptly to antiviral treatment; one patient (sifalimumab 200 mg monthly) experienced a recurrence of Herpes zoster during the study, and one patient (sifalimumab 1200 mg monthly) discontinued because of a Herpes zoster infection. In addition, one patient (sifalimumab 1200 mg monthly) had ophthalmic Herpes zoster noted at day 1, which was successfully treated.
One anaphylactic reaction (nasal congestion, chest discomfort, tachycardia, facial flushing, and swelling of the lips, tongue and vulva in association with dyspnoea and bronchospasm) was reported in a patient receiving placebo, which resolved within 10 min following treatment with intravenous clorprenaline. Hypersensitivity events occurred in two patients receiving placebo, one of whom experienced two events, and three patients on sifalimumab (200 mg, 1200 mg (2)). Of these, one patient (placebo) discontinued treatment. Overall, there were 26 infusion-related reactions with a similar incidence in the placebo (5.6%) and combined sifalimumab (6.2%) groups.
During the course of the study (including follow-up), no clinically important shifts or changes were observed in haematology, chemistry, urinalysis parameters, vital signs, lipid parameters or ECG in any of the treatment groups.
Discussion
This phase IIb study demonstrated greater efficacy with IFN-α pathway blockade than placebo in the treatment of patients with moderate to severe active SLE and an inadequate response to standard-of-care treatments. The broad-based improvement measured in both SLE composite end points (SRI(4), BICLA) and individual organ systems (CLASI, joint counts) supports preclinical and clinical evidence that IFN-α plays an important role in SLE pathogenesis.10 ,34–36 Although the 1200 mg dosage provided the most consistent results, no clear sifalimumab dosage effect was observed in the study. These potentially promising results are important in early drug development, but are not definitive until prospectively replicated in larger studies with a more stringent statistical significance level (eg, α=0.05).
Assessment of SLE manifestations using SLEDAI-2K was less sensitive in detecting improvements than more comprehensive measures of individual organs. In particular, CLASI assessments demonstrated greater sensitivity and rapidity in detecting skin improvements compared with the SLEDAI-2K mucocutaneous system assessments (figure 3), and the post hoc analysis of joint improvements captured greater improvements compared with the SLEDAI-2K musculoskeletal system assessments. Although there was no requirement for patients to have a minimum number of swollen/tender joints for inclusion in this study, the positive findings from the exploratory analysis are encouraging, given the high prevalence of arthritis among patients with lupus.
In the sifalimumab 600 mg group, there appeared to be a slight reduction in CLASI responders, in the subset of patients with baseline CLASI activity score ≥10, after week 28. As no neutralising antibodies to sifalimumab were present in any patient the reason for this decrease is not clear. Larger studies would reduce the relatively high variabilities that are a consequence of the small patient numbers in this subset (CLASI ≥10 at baseline) and provide a more precise estimate of effect. It should be noted that despite a high placebo response rate in the CLASI activity score, the response to sifalimumab was greater than with placebo, with clear separation observed from 2 months. Sensitivity analysis (see online supplementary table S3) at week 52 showed that, overall, 60.9% of patients who received sifalimumab, compared with 40.0% who received placebo, experienced a 50% improvement in CLASI for patients with CLASI activity score ≥10 at baseline.
The greater distinction from placebo seen for the IFN-high patients supports the hypothesis that the peripheral blood IFN test status reflects systemic type I IFN activity. In contrast, for IFN-low patients there was a smaller difference in response rates between the placebo and the 200 mg or 1200 mg monthly sifalimumab groups. This is not due to a reduced response to sifalimumab, but to a greater placebo response rate; the reason for which is unclear.
Although subgroup analysis of SRI(4) by geographical region demonstrated greater response rates to both sifalimumab and placebo in the predefined high standard-of-care response regions (region 1), the discrimination between the sifalimumab and placebo groups was greater in the low standard-of-care response regions (region 2). This was primarily due to a lower response in the placebo group in region 2, and not to a lower response to sifalimumab in region 1. The geographical disparity was not attributable to differences in baseline SLEDAI-2K, PGA, BILAG-2004 scores or baseline IFN gene signature which were similar in both regions; however, it may be a reflection of variation in other baseline demographic or clinical characteristics between the two regions (see online supplementary table S1). Of note, in the placebo group a higher proportion of patients in region 1 were taking corticosteroids compared with region 2, whereas regional differences in use of these medications were less pronounced in patients treated with sifalimumab. The importance of this observation to explain the greater placebo responses seen in region 1 remains to be determined.
Although the effect sizes observed in the overall population were modest, the differences between the placebo and sifalimumab treatment groups were clinically meaningful and similar to those of other biological therapies. Under-representation of patients from region 2 due to lower than anticipated enrolment may have contributed to minimising the overall differences from placebo. Further delineation of these regional anomalies determined from larger studies is warranted.
Adverse events occurred with similar frequencies in the sifalimumab and placebo groups, except for Herpes zoster infection, which was more common with sifalimumab. This is consistent with the mechanism of action of sifalimumab and safety results reported from a previous study.22 Importantly, the clinical course of Herpes zoster infections was uncomplicated in all cases. These infections responded promptly to therapy, with only one recurrence among patients who continued receiving sifalimumab. As modulation of the type I IFN pathway can potentially disrupt mechanisms of viral defence and therefore, increase susceptibility to viral infections or malignancies, additional, larger studies are needed to fully characterise the safety of this treatment and to define those at highest risk of complications.
In summary, this study demonstrated clinical efficacy of inhibiting IFN-α with sifalimumab in SLE. The clinical effects of sifalimumab are supported by improvements in both global and organ-specific outcome measures, with an acceptable safety profile. Our observations demonstrate that blocking the type I IFN pathway is a promising approach for the treatment of moderate to severe active SLE.
Acknowledgments
The authors thank the patients and investigators and their site personnel who contributed to the study. The authors acknowledge Jennifer Buynitzky, MS, MBA, of MedImmune and Michael A Nissen, ELS, of AstraZeneca for their editorial contributions and facilitation of the development of the manuscript, and Mark Hughes, PhD, of QXV Comms, an Ashfield business, part of UDG Healthcare, Macclesfield, UK, who provided medical writing assistance.
References
Supplementary materials
Lay summary
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Tore K Kvien
Contributors Representatives of MedImmune conducted the data analyses. The first draft of this manuscript was written by WG, and all authors interpreted the data and participated in the preparation of the manuscript with support from professional medical writers. All of the authors made the decision to submit the manuscript for publication, and confirm the veracity and completeness of the data and analyses, as well as the ethical conduct and reporting of the study according to its trial protocol.
Funding The study was funded by MedImmune.
Competing interests MK received a grant for this study from MedImmune/AstraZeneca; grants for other work from Bayer; and personal consultancy fees from INOVA Diagnostics, MedImmune, GlaxoSmithKline and UCB. JTM reports personal fees from MedImmune/AstraZeneca; and grants and personal fees from Genentech/Roche outside of the submitted work. VPW received personal fees from MedImmune/AstraZeneca, during the conduct of this study. RF, KK, GGI, JD, LW and WG received personal fees from AstraZeneca/MedImmune. WG and GGI hold stocks in AstraZeneca/MedImmune. JD has a patent pending. VPW is a named inventor and holds a patent for the CLASI copyright owned by the University of Pennsylvania. GGI, JD, LW and WG are employees of MedImmune.
Provenance and peer review Not commissioned; externally peer reviewed.
Ethics approval This study was conducted in accordance with the principles of the Declaration of Helsinki and the International Conference on Harmonisation Guidance for Good Clinical Practice. Independent ethics committee approval was obtained and all patients provided written informed consent in accordance with local requirements.