Article Text

Abstract
Objectives To determine the safety, tolerability and signs of efficacy of MOR103, a human monoclonal antibody to granulocyte–macrophage colony-stimulating factor (GM-CSF), in patients with rheumatoid arthritis (RA).
Methods Patients with active, moderate RA were enrolled in a randomised, multicentre, double-blind, placebo-controlled, dose-escalation trial of intravenous MOR103 (0.3, 1.0 or 1.5 mg/kg) once a week for 4 weeks, with follow-up to 16 weeks. The primary outcome was safety.
Results Of the 96 randomised and treated subjects, 85 completed the trial (n=27, 24, 22 and 23 for pooled placebo and MOR103 0.3, 1.0 and 1.5 mg/kg, respectively). Treatment emergent adverse events (AEs) in the MOR103 groups were mild or moderate in intensity and generally reported at frequencies similar to those in the placebo group. The most common AE was nasopharyngitis. In two cases, AEs were classified as serious because of hospitalisation: paronychia in a placebo subject and pleurisy in a MOR103 0.3 mg/kg subject. Both patients recovered fully. In exploratory efficacy analyses, subjects in the MOR103 1.0 and 1.5 mg/kg groups showed significant improvements in Disease Activity Score-28 scores and joint counts and significantly higher European League Against Rheumatism response rates than subjects receiving placebo. MOR103 1.0 mg/kg was associated with the largest reductions in disease activity parameters.
Conclusions MOR103 was well tolerated and showed preliminary evidence of efficacy in patients with active RA. The data support further investigation of this monoclonal antibody to GM-CSF in RA patients and potentially in those with other immune-mediated inflammatory diseases.
Trial registration number NCT01023256
- Rheumatoid Arthritis
- DMARDs (biologic)
- DAS28
- Treatment
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Despite major advances in the treatment of rheumatoid arthritis (RA), many patients are unable to achieve treatment goals.1 ,2 There is thus a continuing need for the exploration and development of therapeutic strategies with novel mechanisms of action.
One molecule that may play a critical role in inflammatory arthritis is granulocyte–macrophage colony-stimulating factor (GM-CSF). Although originally characterised by its ability to promote myeloid haematopoiesis, GM-CSF is associated with a multitude of additional effects on mature myeloid cells, including stimulation of the production of inflammatory mediators by neutrophils and macrophages3 ,4 and promotion of the differentiation and pathogenicity of proinflammatory T-helper 17 cells.5 ,6
Several lines of data suggest that GM-CSF strongly influences the development and pathogenesis of RA.7 Animal models support a key role for this molecule in both initiating and exacerbating inflammatory arthritis.8–11 In humans, GM-CSF is found at elevated levels in the synovial tissue and fluid of patients with RA.12 ,13 Exacerbation of established RA has been reported in patients who received GM-CSF as supportive therapy.14 ,15 More recently, clinical trials have found that GM-CSF receptor-α blockade reduced disease activity in patients with RA.16 ,17
Targeting the cytokine directly by means of a monoclonal antibody to GM-CSF provides an alternative means of blocking GM-CSF. MOR103 is a high-affinity recombinant human IgG1 antibody that binds to a GM-CSF epitope, thereby blocking cytokine–receptor interaction and receptor activation.18 Although GM-CSF receptor blockade and direct GM-CSF targeting are both expected to block GM-CSF-mediated signalling, the targeting of receptor versus cytokine could potentially result in different target-mediated drug disposition. In addition, since MOR103 targets the soluble cytokine, no antibody- or complement-dependent cytotoxicity is anticipated. We report the results of the first in patient study with MOR103 in patients with RA.
Methods
Trial design and treatment
This trial (NCT01023256) was a randomised, double-blind, placebo-controlled, multidose, dose-escalation trial of three MOR103 doses (0.3, 1.0 and 1.5 mg/kg). These doses were chosen on the basis of a previous safety study in healthy human subjects and pharmacokinetic modelling of trough levels required for GM-CSF inhibition in synovial tissue. Additional information on the study drug manufacturer and intravenous administration can be found in the online supplementary text.
Subject eligibility was determined at the screening visit (up to 35 days before treatment initiation) and confirmed at baseline before the first dose on day 1. Eligible subjects were enrolled into three cohorts according to a randomisation schedule through an interactive web response system. All investigators and participants were blinded to the study randomisation scheme.
Each subject received a total of four doses, one per week at baseline and days 8 (week 1), 15 (week 2) and 22 (week 3). Subjects made follow-up visits to the trial centre at weeks 4, 5, 6, 8, 10, 13 and 16. An independent Data Safety Monitoring Board (DSMB) reviewed an interim safety report with data from at least 20 subjects in each of the first two cohorts (0.3 and 1.0 mg/kg). DSMB approval was required before the study was allowed to proceed to the next higher dose. A second safety review was performed when all subjects in these cohorts had completed their week 5 visit.
The trial was initiated on 19 January 2010 and the last visit was on 14 June 2012. Subjects were treated in 26 centres in Europe (see online supplementary table S1 for participating countries). The trial protocol was approved by the institutional review boards or independent ethics committees at the participating sites and was conducted in accordance with the Declaration of Helsinki (revised edition, Seoul 2008) and the International Conference on Harmonisation Guidelines for Good Clinical Practice. Patients provided written informed consent for the trial before undergoing screening procedures. Authors had full access to data and certify the accuracy and completeness of data analysis.
Subjects
Men or women 18 years of age or older with active RA according to the revised 1987 criteria of the American College of Rheumatology (ACR)19 and a body mass index between 19.0 and 35.0 kg/m2 (inclusive) were eligible for this trial. Subjects were required to have: (1) at least three swollen and three tender joints using the Disease Activity Score-28 joints (DAS28) joint count, with at least one swollen joint in the wrist or hand excluding the proximal interphalangeal joints; (2) C-reactive protein (CRP) levels >5 mg/L if seronegative for rheumatoid factor and anti-cyclic citrullinated peptide antibody, or >2 mg/L if seropositive for either marker; (3) DAS28 ≤5.1; and (4) ACR functional class of I, II or III.20 Patients with high disease activity (DAS28 >5.1) were excluded as the efficacy of MOR103 was unknown. Subjects had to be willing to use effective contraception, be surgically sterile or at least 2 years postmenopausal. Previous treatment with biological/immunosuppressive therapies other than cell-depleting agents was allowed with an adequate washout period. Stable concomitant treatment with non-biological disease-modifying antirheumatic drugs or low doses of oral corticosteroids was allowed. Exclusion criteria were presence or history of major chronic inflammatory autoimmune disease, clinically significant abnormalities in haematology parameters, liver enzymes or pulmonary function tests (PFTs), and significant systemic illness or malignancy.
Safety and tolerability assessments
The primary end point was the evaluation of safety and tolerability. Data on adverse events (AEs; MedDRA V.13.0), vital signs, serum chemistry, haematology and urinalysis were collected at each visit. Additional evaluations included coagulation parameters, whole blood flow cytometry, and ECG. Samples for laboratory analyses were collected before dosing. Laboratory analyses were performed at a central laboratory (Eurofins Medinet BV, Breda, The Netherlands).
Because high levels of GM-CSF autoantibodies have been associated with idiopathic pulmonary alveolar proteinosis,21 ,22 serum surfactant D levels were measured at all visits except the screening visit, and PFTs, including forced expiratory volume in the first second (FEV1), vital capacity, forced vital capacity and diffusing capacity of the lung for carbon monoxide (DLco), were performed at screening and weeks 2, 4, 10 and 16. Decreased DLco was reported as an AE only if judged to be potentially clinically relevant by the treating physician. An additional evaluation of patients experiencing a ≥20% decrease in DLco compared with screening was also conducted.
Serum levels of proinflammatory cytokines, including tumour necrosis factor (TNF), interleukin (IL)-1β, IL-6, IL-8, IL-10, interferon (IFN)-γ and GM-CSF (free cytokine only, not MOR103-bound GM-CSF), were measured at baseline, weeks 1–4 and week 16 by use of the Cytokine Multiplex Kit (Life Technologies GmbH, Darmstadt, Germany) and a Luminex reader. A validated direct ELISA was used to test patient serum samples for MOR103 antibodies at screening and weeks 10, 13 and 16. Bound drug antibodies were detected using anti-IgG- or anti-IgM-specific secondary antibodies.
Clinical assessments
The primary exploratory efficacy outcome was change from baseline in DAS28 calculated using the erythrocyte sedimentation rate as the acute phase reactant.23 Other exploratory efficacy assessments included the proportions of patients achieving European League Against Rheumatism (EULAR) response24 and ACR improvement criteria25 and changes in DAS28 and ACR core measures, including tender joint count (TJC; 69 joints), swollen joint count (SJC; 66 joints), patient's self-assessment of pain (measured on a 100 mm visual analogue scale) and the Health Assessment Questionnaire-Disability Index (HAQ-DI).26 Fatigue was measured at baseline and weeks 4, 8 and 16 by the Functional Assessment of Chronic Illness Therapy (FACIT)-fatigue self-assessment scale, which has been validated in RA.27 MRI of the wrist and hand was performed at screening, week 4 and week 8. The side with a swollen wrist or the most swollen joints within the 2nd to 5th metacarpophalangeal (if neither or both wrists were swollen) was selected for imaging; the right side was chosen if swollen joints were equivalent. Two independent experts blinded to patient data and image chronology scored the images in a single reading campaign using the Outcome Measures in Rheumatology (OMERACT) RA MRI Studies (RAMRIS) scoring system.28 ,29 Central medical imaging services were provided by BioClinica (Newton, Pennsylvania, USA) and its affiliates.
Statistical methods
Data were analysed by descriptive statistics using SAS for Windows, Release V.9.3. The Cochran–Mantel–Haenszel test and Fisher's exact test were used to determine p values for EULAR and ACR response rates, respectively. p Values for other efficacy measures were derived from pairwise comparisons between each dosage group and the pooled placebo group based on an analysis of covariance (ANCOVA) model including fixed-effect terms for dose and the covariate CRP level at baseline. p Values were calculated on outcome parameters from weeks 4 through 16 but not at earlier visits, as the efficacy outcome of greatest interest was response after completion of treatment. Post hoc analyses of differences in baseline demographic characteristics among groups used the Kruskal–Wallis test. p<0.05 indicated significance.
To minimise the risk of exposing a large number of subjects without evidence of clinical benefit, sample size calculations were based on the primary exploratory efficacy outcome, change in DAS28. Assuming a statistical power of 80%, an α of 0.05 (two-sided test), a common SD of 1.0, and a discontinuation rate of 15%, we estimated that 21 subjects were required for each MOR103 treatment group and seven subjects for each placebo group (pooled placebo group of 21) to demonstrate a difference of 1.0 point in mean DAS28 change from baseline in the active group compared with placebo. This treatment difference was based on the DAS28 difference between active and placebo treatments of 1.3–1.6 observed over 12 weeks with adalimumab,30 adjusted for length of exposure.
Results
Subjects
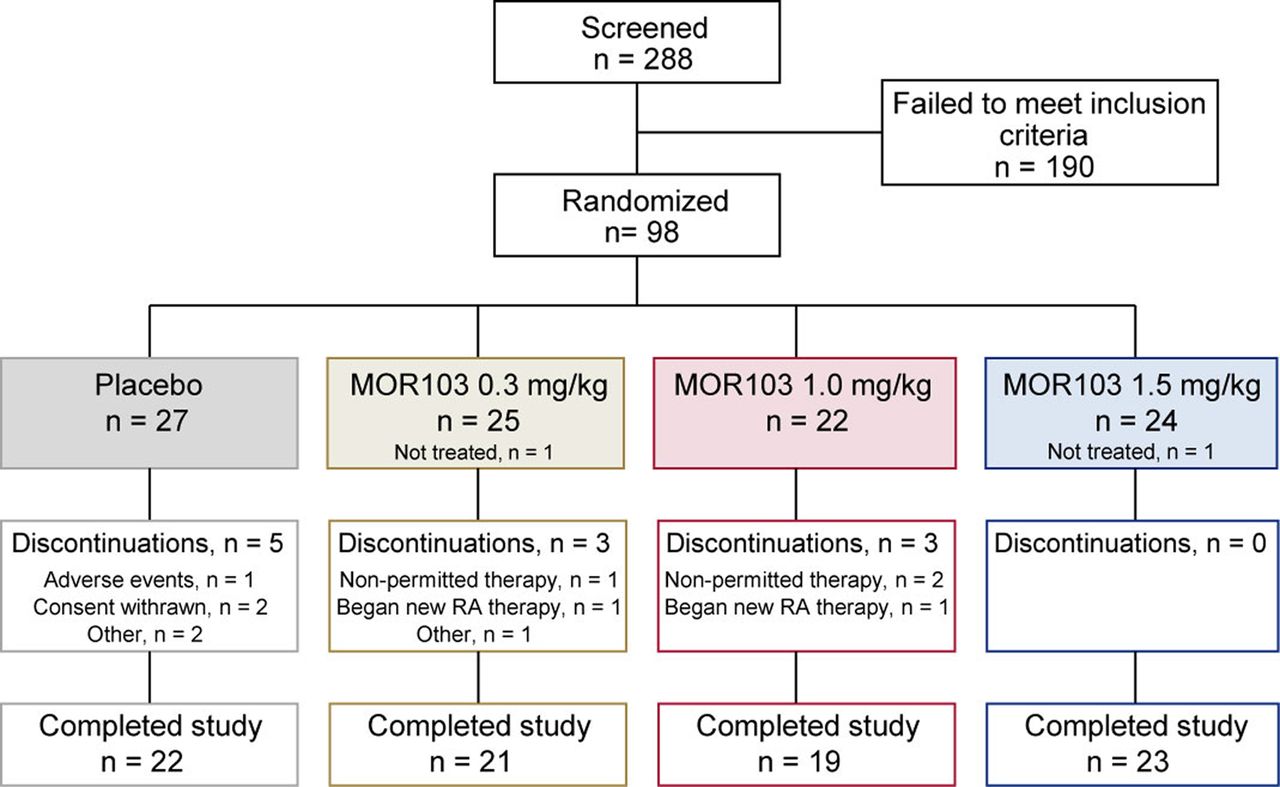
Of the 288 subjects who were screened, 98 were randomised, 96 received treatment at 26 trial centres in Europe, and 85 (86.7%) completed the study (figure 1; see online supplementary table S1 for distribution by country). Two randomised patients did not receive treatment and were excluded from all analyses. All patients who received treatment (N=96; full analysis set) were included in outcome assessments.

Disposition of patients with rheumatoid arthritis randomised to receive placebo or MOR103 in the full analysis set.
Baseline demographic and clinical characteristics were generally comparable among treatment groups, although post hoc analyses revealed differences in mean CRP levels and TJC that reached the level of significance (table 1).
Baseline demographic and clinical characteristics
Safety and tolerability
A total of 144 treatment-emergent AEs were reported in 54 (56.3%) subjects (42 subjects (60.9%) in the MOR103 groups (see online supplementary table S2) and 12 (44.4%) in the pooled placebo group). The most common treatment-emergent AE by preferred term in the active and placebo groups was nasopharyngitis (table 2). The incidences of fatigue, cough and AEs related to RA (worsening or flares) in the MOR103 group were >4% higher than in the placebo group. In eight of the nine MOR103 subjects who reported RA exacerbations, this AE occurred after the last dose of MOR103 (10 days to >12 weeks), suggesting that disease flares were related to withdrawal of active treatment. No cases of infusion reaction were reported, but temporal correlations suggest that one case of rash (MOR103 0.3 mg/kg) and one of fatigue (MOR103 1.0 mg/kg) may have been related to MOR103 infusion. Nasopharyngitis, fatigue, RA exacerbations, rhinitis and oropharyngeal pain occurred at higher incidences (>4% higher) in the MOR103 1.0 mg/kg group than in other active treatment groups or the pooled placebo group. These differences should be interpreted with caution as they are based on small subject numbers.
Incidence of treatment-emergent adverse events occurring in >2 subjects in any treatment group
None of the AEs were considered to be probably or definitely related to treatment. AEs possibly related to treatment were reported in seven placebo (14 AEs) and 10 MOR103 subjects (19 AEs; table 3). Only three AEs (fatigue, anaemia and decreased DLco) were considered possibly related to treatment in more than one subject.
Incidence of possibly treatment-related AEs by preferred term for AEs occurring in one or more MOR103-treated subjects
All AEs were judged to be of mild or moderate intensity except for one severe AE of hospitalisation due to paronychia in the placebo group. The patient recovered fully. There were two serious treatment-emergent AEs during the study: the aforementioned case of paronychia (placebo group) and pleurisy of moderate intensity (MOR103 0.3 mg/kg group), both of which resulted in hospitalisation. In the MOR103 subject, pleurisy was detected 5 days after the last dose of the trial. The patient was treated with antibiotics and recovered fully. One treatment-emergent AE in a placebo patient, decreased DLco, resulted in treatment discontinuation. There were no deaths in the trial.
Additional evaluations of subjects with a ≥20% decrease in DLco or alterations in PFTs did not reveal any patterns of concern in MOR103-treated patients (see online supplementary text). There were no clinically important changes in surfactant D levels, vital signs, ECG, whole blood flow cytometry or urine, haematology or serum chemistry parameters during treatment and no clinically important differences between active treatment and placebo in these measures.
None of the peripheral blood cytokines evaluated in this trial showed notable or consistent changes related to MOR103 infusions (data not shown). The levels of GM-CSF and certain other cytokines, including IFNγ, TNF and IL-10, remained at or near the lower limit of quantification throughout the trial.
Serum samples were tested for anti-MOR103 IgG and IgM antibodies. Only sporadic positives were detected (see online supplementary text), and there was no evidence that antibodies to MOR103 affected serum MOR103 concentrations or clinical outcomes.
Efficacy
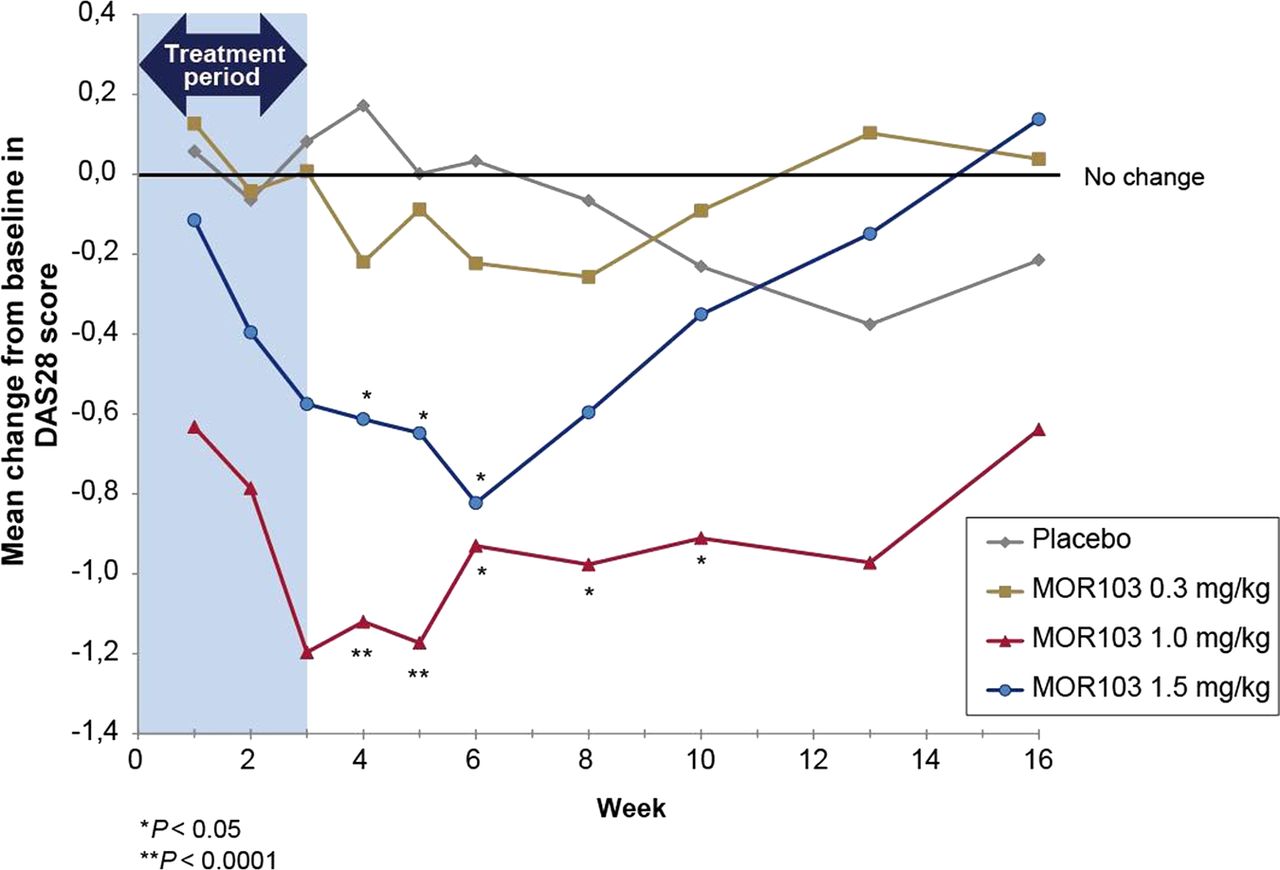
In exploratory investigations, the two higher doses of MOR103 were associated with significant reductions in disease activity. Compared with the pooled placebo group, significantly greater changes from baseline in DAS28 scores (the primary efficacy outcome according to study protocol) were observed in the MOR103 1.0 mg/kg group from weeks 4 through 10 and in the MOR103 1.5 mg/kg group from weeks 4 through 6 (figure 2; see online supplementary figure S1 for mean DAS28 scores during treatment). There were no significant differences in DAS28 changes between placebo and MOR 103 0.3 mg/kg. In the MOR103 1.0 and 1.5 mg/kg groups, improvements in DAS28 scores were observed at the earliest post-treatment visit (week 1). By week 16, mean DAS28 scores were at or above baseline in all groups except MOR103 1.0 mg/kg, which continued to maintain DAS28 scores at levels well below baseline. As with DAS28, the MOR103 1.0 mg/kg group showed the most pronounced effects in additional efficacy outcomes (highest response rates and largest reductions in disease activity parameters; table 4).
Efficacy outcomes at weeks 4 and 8

{kind=link}
{kind=link}
Mean change from baseline in DAS28 scores. Statistical significance was not evaluated before the week 4 visit as specified in the study protocol. DAS28, Disease Activity Score-28 joints.
MRIs were evaluated for synovitis, bone oedema and erosions at weeks 4 and 8 by the OMERACT RAMRIS scoring system. No statistically significant differences between the pooled placebo group and any of the MOR103 groups were observed (data not shown). The largest difference between active treatment and placebo was for the MOR103 1.0 mg/kg synovitis score at week 4 (−0.84 vs placebo; p=0.35).
Discussion
A growing body of literature has documented the potential role of GM-CSF in the pathogenesis of RA. Here we present the first in patient data on the effects of GM-CSF blockade by means of a fully-human antibody to GM-CSF, MOR103, in patients with RA. Although the primary objective of this trial was to determine the safety and tolerability of multiple doses of MOR103, exploratory analyses provided suggestive evidence for the efficacy of this agent.
In this phase Ib/IIa clinical trial, MOR103 was well tolerated and associated with a satisfactory safety profile in patients with active moderate RA who received up to four doses at weekly intervals. Although overall rates of treatment-emergent AEs were higher in the MOR103 groups (60.0%) compared with placebo (44.4%), most treatment-emergent AEs occurred at similar incidences. Fatigue, cough and RA worsening/flares (primarily occurring after the end of active treatment) were reported more frequently by MOR103 subjects than placebo subjects. All treatment-emergent AEs in the MOR103 groups were of mild or moderate intensity. One serious AE, pleurisy of moderate intensity, occurred in a MOR103 subject (0.3 mg/kg) and resolved on antibiotic treatment.
Because of the association between GM-CSF autoantibodies and idiopathic pulmonary alveolar proteinosis,21 ,22 surfactant D levels and PFTs were closely monitored. Decreased DLco was observed in some subjects, but there were no obvious differences between placebo and active treatment groups. Surfactant D levels remained normal and there was no overall pattern of PFT alterations in MOR103-treated subjects. Similar findings on the lung safety of anti-GM-CSF receptor therapy were reported in the phase II mavrilimumab trial.17 Cytokine release was not observed in MOR103-treated subjects. Anti-MOR103 antibody testing detected only sporadic positives, and there was no clear association between assay results and clinical outcomes.
In exploratory efficacy analyses, MOR103 resulted in significant improvements relative to placebo in several outcome measures. The most pronounced effects were observed with MOR103 1.0 mg/kg. The effect of this dose on disease activity was robust for all major efficacy variables and over time, and did not appear to be driven by geographical imbalance, individual centres, concomitant medications, or outliers. Subjects treated with MOR103 1.5 mg/kg also showed significant improvements in certain efficacy parameters, but the changes were not as pronounced or as sustained as for MOR103 1.0 mg/kg. A rapid onset of action was observed at both of the higher MOR103 doses. MOR103 0.3 mg/kg resulted in significant improvements over placebo in SJC at week 4, but did not affect other measures of disease activity.
It seems unlikely that the discrepancy in clinical activity between the 1.0 and 1.5 mg/kg doses reflects a true difference in clinical efficacy, as the difference in dose is relatively small and we are not aware of other studies of cytokine inhibition with a bell-shaped optimum response curve. As for other phase I/II clinical trials, limitations of this study include its small sample size, limited duration, and exclusion of patients with severe RA. Larger clinical trials are needed to define the optimal MOR103 dosage.
It is too early to comment on possible differences in clinical profiles between MOR103 and mavrilimumab, an antibody to GM-CSF receptor.16 ,17 Because of the different targets of these two agents (soluble cytokine vs membrane-bound receptor), there may be differences in tolerability and spectrum of activity. Further studies will be required to explore these issues.
The data presented here establish proof of concept for the use of antibodies to GM-CSF in the treatment of RA and support the initiation of larger clinical trials to confirm the safety and efficacy of MOR103. Our findings suggest that MOR103 has the potential to become a novel and valuable therapeutic option for RA.
Acknowledgments
We thank all of the patients and investigators who participated in this trial. We also thank Samson Fung (Fung Consulting, Eching, Germany) and Dominika Weinelt (MorphoSys AG, Martinsried/Planegg, Germany) for contributing to data analysis and interpretation, Steffen Stürzebecher (MorphoSys AG, Martinsried/Planegg, Germany) for reviewing the manuscript, and Sharon L Cross, PhD (Mission Viejo, CA) for providing medical writing services on behalf of MorphoSys AG.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
AJS and HB contributed equally.
Contributors All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version. FB, PPT, MØ, TWH, AJS and HB were involved in the study conception and design, FB, PPT, MØ, BJE, RS, PW, TWH, VYB, SV, JR, AR-R, MK, DR, IAZ, BOE, JG, JF and HB were responsible for data acquisition, and FB, PPT, MØ, RPK, AJS and HB were involved in analysis and interpretation of data.
Funding The study was supported by MorphoSys AG, which provided funding for the trial, data analyses, and medical writing services.
Competing interests FB, PPT, MØ, RS, PW, TWH, VYB, SV, JR, AR-R, MK, DR, IAZ and HB have received investigator grants and/or advisory fees from MorphoSys AG, the sponsor of this study. MØ, BJE, JG and JF received compensation from MorphoSys for their work as central readers for MRI or pulmonary function tests during the trial. RPK and AJS are employees of MorphoSys AG.
Patient consent Obtained.
Ethics approval Institutional review boards or independent ethics committees at the participating sites (26 centres in Europe).
Provenance and peer review Not commissioned; externally peer reviewed.