Article Text

Abstract
Objectives We aimed to quantify the risk of major adverse cardiovascular events (MACE) among patients with psoriatic arthritis (PsA), rheumatoid arthritis (RA) and psoriasis without known PsA compared with the general population after adjusting for traditional cardiovascular risk factors.
Methods A population-based longitudinal cohort study from 1994 to 2010 was performed in The Health Improvement Network (THIN), a primary care medical record database in the UK. Patients aged 18–89 years of age with PsA, RA or psoriasis were included. Up to 10 unexposed controls matched on practice and index date were selected for each patient with PsA. Outcomes included cardiovascular death, myocardial infarction, cerebrovascular accidents and the composite outcome (MACE). Cox proportional hazards models were used to calculate the HRs for each outcome adjusted for traditional risk factors. A priori, we hypothesised an interaction between disease status and disease-modifying antirheumatic drug (DMARD) use.
Results Patients with PsA (N=8706), RA (N=41 752), psoriasis (N=138 424) and unexposed controls (N=81 573) were identified. After adjustment for traditional risk factors, the risk of MACE was higher in patients with PsA not prescribed a DMARD (HR 1.24, 95% CI 1.03 to 1.49), patients with RA (No DMARD: HR 1.39, 95% CI 1.28 to 1.50, DMARD: HR 1.58, 95% CI 1.46 to 1.70), patients with psoriasis not prescribed a DMARD (HR 1.08, 95% CI 1.02 to 1.15) and patients with severe psoriasis (DMARD users: HR 1.42, 95% CI 1.17 to 1.73).
Conclusions Cardiovascular risk should be addressed with all patients affected by psoriasis, PsA or RA.
- Psoriatic Arthritis
- Rheumatoid Arthritis
- Outcomes research
- Epidemiology
Statistics from Altmetric.com
Introduction
Psoriatic arthritis (PsA) is a chronic inflammatory arthritis that occurs in approximately 8%–30% of patients with psoriasis.1 ,2 PsA has been linked to an increased prevalence of cardiovascular (CV) comorbidities and CV risk factors.3 However, the majority of studies performed to date have been cross-sectional. Cohort studies examining the risk of incident CV events in PsA are sparse.4 Three population-based studies have examined the risk of CV events among patients with psoriasis and have included patients with PsA as a subgroup.5–7 Existing studies have not examined the risk for incident major adverse cardiovascular events (MACE) including myocardial infarction (MI), cerebrovascular accidents (CVA) and CV death in PsA compared with matched internal controls from a population-based perspective after accounting for the presence of traditional CV risk factors.
Rheumatoid arthritis (RA) and severe psoriasis have been consistently linked to an increased risk for incident MACE.3 ,4 ,8–16 It has been suggested that patients with PsA have a similarly elevated risk for CV disease. However, we recently demonstrated that patients with PsA did not have an increased risk of mortality compared with internal controls, while patients with severe psoriasis (defined as patients with psoriasis prescribed systemic therapy or phototherapy) and RA had substantially elevated mortality (HR 1.75 and 1.54–1.59, respectively).17 This led us to question whether PsA is associated with incident CV disease from a population-based perspective.
The objective of this study was to examine the risk of incident MACE including MI, CVA and CV death controlling for traditional CV risk factors among patients with PsA, RA or psoriasis compared with unexposed controls using a population-based cohort. We hypothesised similar rates of CV disease among the three groups given known associations with systemic Th1-driven and Th17-driven inflammation.18 ,19
Methods
Study design and setting
A cohort study was performed using data from The Health Improvement Network (THIN) in the UK between 1994 and 2010. THIN is a large medical record database in which general practitioners (GP) record routine health data about their patients.20–22 The UK is an ideal setting for examining long-term health outcomes given the gatekeeper model, meaning that GPs are responsible for coordinating all of the patient's care. Additionally, pay-for-performance measures mandate collection of data on CV outcomes and several key CV risk factors including diabetes and smoking.23
Study population
All patients with PsA, psoriasis or RA, and between the ages of 18 and 89 years at the index date were included if they had observation time in THIN after Vision software implementation. Patients were excluded if they had died or were transferred out of the practice prior to the implementation of Vision software. Up to 10 unexposed controls from the general population without PsA, psoriasis, RA, or disease-modifying antirheumatic drugs (DMARD) prescriptions were randomly selected for each patient with PsA and were matched on practice and index date within the practice (defined as latest of registration with the practice and diagnosis date). Unexposed controls were assigned a ‘diagnosis date’ within 6 months of diagnosis date of the patient with PsA. This algorithm was designed to minimise bias by ensuring that PsA and unexposed controls are followed by similar doctors during similar time periods. For each individual outcome analysis, patients were excluded if they had the outcome of interest prior to the index date.
Exposure definitions
PsA, psoriasis and RA were defined by the presence of at least one READ code consistent with these diseases (READ codes are standard medical diagnosis codes used in the UK general practice system).24 READ codes for psoriasis (positive predictive value (PPV) 90%),25 RA (PPV 81% for ‘potential cases’ defined as single code without DMARD, rheumatoid factor result or rheumatology referral)26–28 and PsA (PPV 85%)2 ,29 have been previously validated within the same or analogous large medical record databases. We have used this definition of PsA in other studies.2 ,17 ,30 ,31 Patients were classified as PsA if they had a code for PsA, RA if they had a code for RA but not PsA, and psoriasis if they had a code for psoriasis but did not have a code for RA or PsA.
Outcome definitions
Outcomes were defined by READ codes representing the outcome of interest within the study period. The censoring date was the first occurrence of the outcome of interest. Patients were excluded from each analysis if they had the event of interest prior to the index date. MI and stroke were defined by a previously validated set of READ codes with PPV 93%32 and 77.5%–89.3%, respectively.33 ,34 CV death was defined by a set of READ codes chosen based on the UK ICD10 codes classifying a CV death and the Centers for Disease Control ICD9 codes classifying death as heart disease or stroke.35 ,36 These codes were extracted within the 60 days before, or earlier than 180 days, following a code signifying the patient's death. Text comments in the database reporting the patient's death as CV were also used to classify CV death. This algorithm has been used previously and is the recommended method for identifying cause of death by THIN. MACE, the composite outcome, was achieved at the first of MI, CVA or CV death.
Person time calculation
The index date (cohort entry) was defined as the latest date of the following events: diagnosis date, 6 months after initial registration with the practice, DMARD initiation (in patients using DMARDs), implementation of Vision software in the patient's practice or a practice-acceptable mortality reporting.37–40 The index data was similarly calculated in unexposed controls except for ‘diagnosis date’ was the diagnosis date of the matched patient with disease. Censoring occurred when the event of interest occurred, the patient died, the patient left the practice, the practice stopped participating in THIN, or the study ended in September 2010.
Covariates of interest
All covariates of interest were measured prior to the index date. A priori, we hypothesised a statistical interaction between disease status and both age and DMARD use, as disease severity may be reflected by DMARD use. DMARD exposure was included in the models as a binary variable for exposure at any point up to the index date. Among patients with RA and PsA, DMARDs included methotrexate, sulfasalazine, azathioprine, leflunamide, cyclosporine, mycophenolate, hydroxychloroquine; and biological disease-modifying agents including adalimumab, etanercept and infliximab. In patients with skin psoriasis without a diagnosis of PsA or RA, methotrexate, ciclosporin, biological disease-modifying agents, phototherapy, psoralen plus ultraviolet light therapy (PUVA), retinoids (acitretin and etretinate) and hydroxyurea were considered DMARDs. In the UK, DMARDs can be prescribed by consultants (specialists) but should be captured by GP records with the exception of the biological medications, which are rarely recorded.20 The following potential confounders were measured: age, sex, smoking, body mass index, blood pressure at baseline, year of cohort entry, Townsend Deprivation Score correlated with socioeconomic status,20 urban versus rural living environment, chronic kidney disease, diabetes, hypertension, use of prescription non-steroidal anti-inflammatory drugs (NSAID) and oral corticosteroids prior to index date. The Charlson Comorbidity Score41 was also calculated, but the typical point for RA was not included in order to better capture differences in other comorbidities among the groups.
Statistical analysis
Covariate distribution among the groups was examined using descriptive statistics. Cox proportional hazards regression models were used to calculate the HR for each group compared with the unexposed group after adjusting for age and sex. Hypothesised effect modifiers, use of DMARDs and age, were tested in the models, and the likelihood ratio test was used to determine significance of the interactions. We then tested the hypothesised confounders in the model using a purposeful selection modelling approach42 and kept in the model the predetermined confounders (age, sex and traditional CV risk factors) and covariates that changed the main effects by >10% and had a p value<0.1. Log-log survival plots and Schoenfeld residuals were used to assess the assumption of proportionality of hazards. Several sensitivity analyses were performed (more details given in online supplementary figure S1). Statistical analysis was performed using Stata V.13.0 (College Station, Texas, USA).
Sample size determination
Power calculations prior to the start of the study revealed that with 7000 patients with PsA and 35 000 unexposed patients, we would have 90% power to detect an HR as small as 1.28, 1.16 and 1.19 for CV death, MI and stroke, respectively, with an average of 5 years of follow-up per patient in an unadjusted analysis. Baseline event rates were assumed to be 0.16%, 0.49% and 0.35% per year for CV death, MI and stroke, respectively.
Ethics board approval
All data in this study was anonymous to the investigators. This study was approved by the University of Pennsylvania Institutional Review Board and Cegedim's Scientific Review Committee. This manuscript was prepared according to the Strengthening the Reporting of Observational Studies in Epidemiology statement recommendations.43
Results
Among 8706 patients with PsA, 41 752 patients with RA, 138 424 patients with psoriasis and 82 258 randomly selected unexposed patients meeting the inclusion criteria, follow-up time in the study period was comparable. Baseline characteristics are found in table 1 (additional patient characteristics are found in online supplementary table S1). Patients with RA were older and more often women. Approximately half the patients with RA and PsA were prescribed a DMARD and 3% of patients with psoriasis had been prescribed a DMARD or received phototherapy. At least 65% of patients with PsA and RA had been prescribed NSAIDs compared with 24% with psoriasis and 47% of controls. Compared with the unexposed population, the prevalence of CV risk factors, MI and stroke in the baseline period were elevated in patients with PsA, RA and psoriasis. Reasons for leaving the cohort (censoring) other than having an outcome of interest were similar among groups (data not shown).
Baseline characteristics
The unadjusted incidence rates of MI, CVA and MACE (composite outcome) are reported in table 2. HRs for MI, stroke, CV death and MACE are presented in table 3. There was a significant interaction between DMARD status (ever vs never prescribed) and exposure (disease) group (p<0.001 for CV death, CVA and MACE, and p=0.01 for MI). Therefore, the stratified results are presented. The risk of MACE (composite outcome) was elevated in patients with PsA without a DMARD prescription (HR 1.24, 95% CI 1.03 to 1.49), RA (No DMARD: HR 1.39, 95% CI 1.28 to 1.50 and DMARD user: 1.58, 95% CI 1.46 to 1.70) and severe psoriasis (defined as patients prescribed a DMARD; HR 1.42, 95% CI 1.17 to 1.73).
Incidence rate of cardiovascular death, MI, stroke and MACE
HRs and 95% CIs for major adverse cardiovascular events
Patients with PsA had an elevated risk for incident MI (HR 1.36, 95% CI 1.04 to 1.77 and HR 1.36, 95% CI 1.01 to 1.84 for no DMARD and DMARD, respectively). The risk for MI was similarly elevated in patients with RA without a DMARD prescription (HR 1.33, 95% CI 1.17 to 1.51) and patients with severe psoriasis (HR 1.26, 95% CI 0.92 to 1.72), but was substantially higher in patients with RA who had been prescribed a DMARD (HR 1.96, 95% CI 1.75 to 2.19).
The risk of incident stroke was also significantly elevated in patients with PsA without a DMARD prescription (HR 1.33, 95% CI 1.03 to 1.71) which was similar to patients with RA and severe psoriasis. Finally, CV death was only significantly elevated in RA (no DMARD: HR 1.43, 95% CI 1.28 to 1.59 and DMARD: HR 1.66, 95% CI 1.48 to 1.86) and severe psoriasis (HR 1.54, 95% CI 1.15 to 2.05).
A third interaction with age as a continuous variable was tested and found to be significant (p<0.001 for all four outcomes). The three-way interactions are presented in figure 1. The relative risk is highest in the younger age groups where the absolute risk is low. Few events occurred in patients younger than 50 years of age (13% of MI, 8% of stroke, 3% of CV death and 10% of composite outcomes).
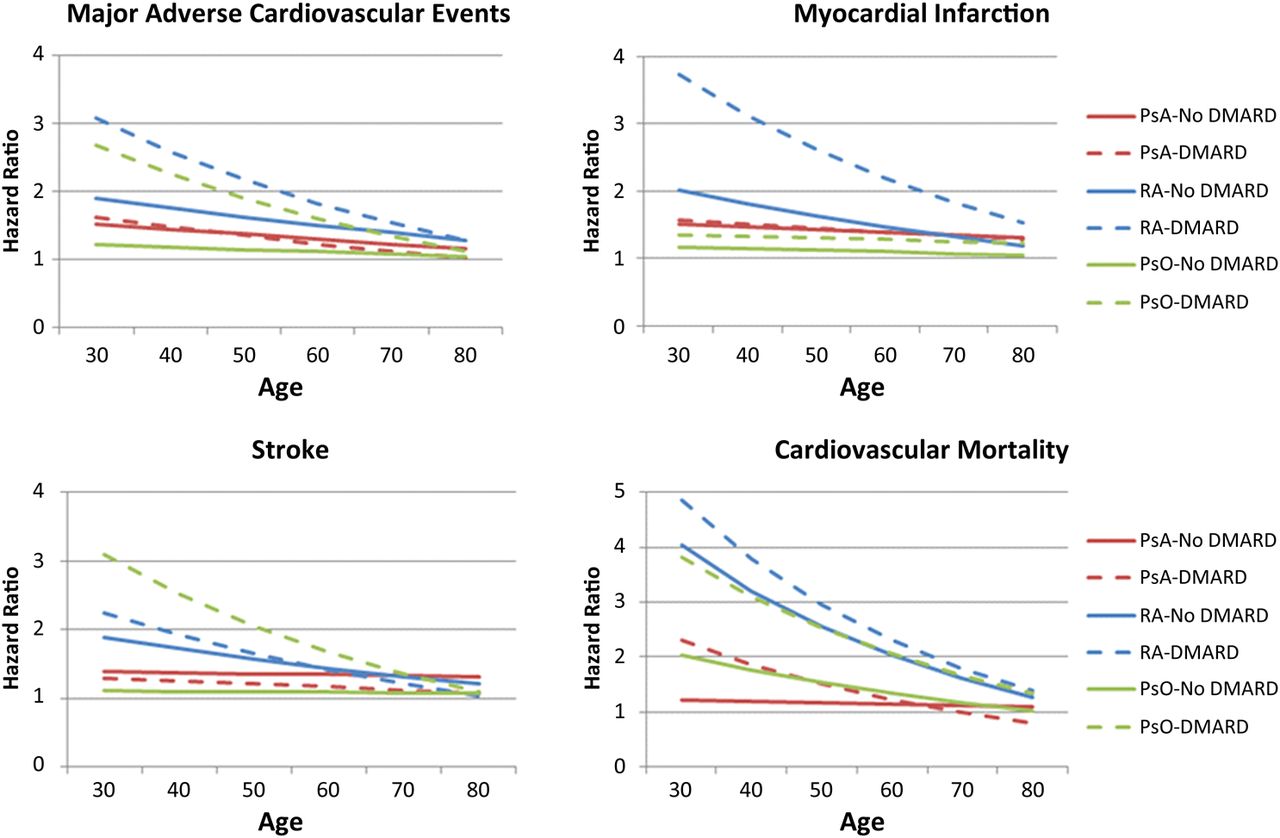
{kind=link}
HRs by age. These graphs incorporate the age interaction into the fully adjusted models for major adverse cardiovascular events, cardiovascular mortality, myocardial infarction and stroke. The fully adjusted models include age, sex, hypertension, diabetes, hyperlipidaemia and smoking status (never, past, current).
Our results were robust to several sensitivity analyses (see online supplementary figure S1); varying definitions of the outcomes, restricting to only patients followed regularly, using multiple imputation for smoking and body mass index, and imputing additional DMARD users. However, in examining the role of death as a competing risk factor for CV events, all previously significant associations in PsA were null, whereas, the HR in the other groups remained unchanged. Finally, adjusting for potentially CV-protective medication use (eg, antihypertensives, lipid-lowering medications and antiplatelet agents listed in online supplementary table S1) during the 1 year prior to start date in the cohort and healthcare use in the baseline period (number of GP visits) did not significantly change the results. One such model is illustrated in online supplementary table S2.
Discussion
To our knowledge, this is the first population-based study dedicated to examining MACE in PsA which may be an independent risk factor for major CV events including MI and stroke, although this was only statistically significant for patients who were not prescribed a DMARD. Additionally, this is the first longitudinal population-based study dedicated to the simultaneous examination of the incidence of MACE in PsA, psoriasis and RA after adjusting for traditional CV risk factors. All three diseases had statistically similar risks for the development of incident CV events after adjustment for age, sex, calendar year of cohort entry and traditional CV risk factors.
Strengths of this study include the large cohort of patients, an average of 5 years of follow-up, simultaneous comparison among three disease cohorts in a population-based study and the use of THIN in which the exposures (psoriasis, RA, PsA) and outcomes (MI, CVA) have been validated. The incidences of MI and CVA in our unexposed population are similar to UK National Statistics,44 lending credence to our algorithms to identify these outcomes and the validity of our unexposed population. These statistics are based on inpatient hospitalisations but support our assumption that we have captured the majority of the outcomes of interest. Furthermore, the increased risk of CV disease in RA and psoriasis are similar to those reported in recent meta-analyses, lending internal validity to our results in PsA.14 ,16
Our study has limitations, including lack of disease activity measures in THIN, generally absent biological medication records, possible missing DMARD prescriptions and the inability to account for over-the-counter NSAID use. THIN does not include data on disease activity in psoriasis or inflammatory arthritis, limiting our ability to examine the effect of disease severity on the incidence of MACE. However, we have shown that simple GP categorisation of body surface area affected by psoriasis is positively correlated with the prevalence of atherosclerotic disease in a prospective study we are conducting nested within the THIN population.45 Use of a systemic DMARD or phototherapy in patients with psoriasis has previously been used as a proxy for severe psoriasis.5 ,10 ,46 However, DMARDs are less likely to represent a pure marker of disease severity in patients with PsA or RA due to confounding by indication and a potential healthy-user effect in patients with PsA (ie, fewer comorbidities in patients with PsA and psoriasis prescribed a DMARD). By contrast, patients with RA prescribed a DMARD could have had more events because their disease was more severe. However, we are unable to test this hypothesis. In patients with PsA who were prescribed a DMARD, the point estimates were nearly the same as patients without a DMARD prescription, but the CI crossed 1.0. This may be due to a lack of statistical power after stratification by DMARD status. It could also be the result of a healthy-user effect, the anti-inflammatory effect of medications on atherosclerosis or closer follow-up in patients using DMARDs, with more attention to CV risk reduction given more frequent physicians visits.47 ,48
NSAIDs have been associated with the development of CV disease,49 although this is less clear in patients with RA.12 Over-the-counter NSAID use may be prevalent among our arthritis cohorts, particularly given that the majority of patients with arthritis have received a prescription NSAID. This should not substantially affect our results, however, as adjustment for prescription NSAID use did not change the main effects (results not shown). Similarly, GPs often do not record the use of biological DMARDs, as these are prescribed directly by rheumatology consultants in the hospital setting.20 However, according to National Institute for Health and Care Excellence guidelines in the UK, all patients must first fail at least one oral DMARD in order to receive a biological DMARD prescription,50 so these patients should have been captured in the ‘DMARD’ group. However, in some cases, the rheumatology consultant will directly prescribe an oral DMARD and the GP may not record this. In a recent validation study, we examined the agreement between GP and medical record report of DMARD use and found that while agreement is overall good, 20 of 53 (38%) patients without a code for a DMARD were reported by the GP to have received a DMARD at some point. In the study, a total of 51 of 87 patients (59%) had either a code for a DMARD or GP report of DMARD use.29 Therefore, in a sensitivity analysis, we augmented the number of DMARD users by first deriving a propensity score (a treatment prediction model) and then assigning those in the top three quintiles a DMARD prescription; this did not change the results. Finally, there may be patients with PsA in the ‘psoriasis-only’ cohort who have not yet been diagnosed with inflammatory arthritis or whose diagnosis was not recorded. This concern is not unique to population-based studies but a general issue that makes comparison of cohorts of patients with psoriasis and PsA challenging.51 The goal of this study was to examine PsA with high specificity and without physical examination or direct questioning of the patients; we did not seek to identify patients with subclinical or undiagnosed PsA.
In conclusion, we report an increased incidence of MACEs in PsA, psoriasis and RA. The HRs for RA and psoriasis were similar to risk estimates in previous studies providing internal validity for the study results in patients with PsA and external validity for the study as a whole. These results suggest the need for improved screening and management of traditional CV risk factors in patients with inflammatory diseases. Future, prospective, randomised, controlled studies are needed to better understand the impact of systemic therapy in decreasing the risk of MACEs in these diseases. Additionally, studies addressing the impact of interventions for traditional CV risk factors on reducing the risk for MACE in patients with inflammatory diseases are needed.
Acknowledgments
We would like to thank Peter Merkel, MD, MPH for helpful discussions and Ronac Mamtani, MD MSCE, James Floury MD MSCE, and Joy Wan, MD, for assistance in assembling code lists.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Lay summary
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Footnotes
Handling editor Tore K Kvien
AO and YDY are co-first authors.
Contributors AO and JMG conceptualised and designed the study with input from YDY, NNM, KH, SH, ABT, TJL and HC. SM and YJ assisted in assignment and analysis of cause of death. All authors were integral in interpretation of the results. AO and YDY performed the programming, statistical analysis, preparation of the data and the first draft of the manuscript. KH performed data abstraction from The Health Improvement Network and assisted in programming. All authors were involved in critical review of the data as well as drafting and revision of the manuscript, and all have approved the final version of the paper to be published.
Funding This project was funded by the American College of Rheumatology (AO), R01HL089744 (JMG), K24AR064310 (JMG). Data from The Health Improvement Network is supported by the Clinical and Translational Science Award at the University of Pennsylvania (8UL1TR000003 from the National Center for Research Resources). AO was supported by the American College of Rheumatology Research and Education Foundation and is now supported by NIH K23AR063764. YY was supported by a grant from the Doris Duke Charitable Foundation and a grant from the Center for Clinical Epidemiology and Biostatistics. SH was supported by R01AG025152. TJL was supported by The Icelandic Research Fund, #120433021. NNM was supported by K23HL097151-01. This work was completed independent of the funders.
Competing interests JMG serves as a consultant to Amgen, Abbott, Centocor, Celgene, Novartis, Eli Lily and Pfizer and has received honoraria. He has received grants from Amgen, Abbott, Pfizer, Novartis, Eli Lily and Genentech. KH and SH are supported by sponsored research agreement between the University of Pennsylvania and Astra Zeneca and Bristol Myers Squibb. SH has consulted for Bristol-Myers Squibb, AstraZeneca and Bayer Healthcare, and has received institutional support for pharmacoepidemiology training from Pfizer. Cegedim Strategic Data (CSD) Medical Research UK is an expert in UK anonymous patient data for the healthcare industry. CSD is a commercial organisation that supplies data and trains and supports researchers in the use of primary care patient data. Data are available for use in medical research in the academic setting as well as in industry for a fee which varies depending on the type of data requested. Aside from undergoing ethical review by the Scientific Review Committee at Cegedim, independent academic groups who voluntarily act as an ethical review body, this protocol was not in any way discussed with Cegedim, nor were any changes made by the company. We did not receive financial support or other forms of computational or analytical support from Cegedim/THIN. The data were collected by Cegedim and the general practitioners without knowledge of the study objectives and hypotheses.
Provenance and peer review Not commissioned; externally peer reviewed.
Data sharing statement Code lists and statistical coding relevant to the manuscript are available from the authors on request.