Article Text

Abstract
Objective This study investigated whether chondrocytes within the cartilage matrix have the capacity to communicate through intercellular connections mediated by voltage-gated gap junction (GJ) channels.
Methods Frozen cartilage samples were used for immunofluorescence and immunohistochemistry assays. Samples were embedded in cacodylate buffer before dehydration for scanning electron microscopy. Co-immunoprecipitation experiments and mass spectrometry (MS) were performed to identify proteins that interact with the C-terminal end of Cx43. GJ communication was studied through in situ electroporation, electrophysiology and dye injection experiments. A transwell layered culture system and MS were used to identify and quantify transferred amino acids.
Results Microscopic images revealed the presence of multiple cellular projections connecting chondrocytes within the matrix. These projections were between 5 and 150 µm in length. MS data analysis indicated that the C-terminus of Cx43 interacts with several cytoskeletal proteins implicated in Cx trafficking and GJ assembly, including α-tubulin and β-tubulin, actin, and vinculin. Electrophysiology experiments demonstrated that 12-mer oligonucleotides could be transferred between chondrocytes within 12 min after injection. Glucose was homogeneously distributed within 22 and 35 min. No transfer was detected when glucose was electroporated into A549 cells, which have no GJs. Transwell layered culture systems coupled with MS analysis revealed connexins can mediate the transfer of L-lysine and L-arginine between chondrocytes.
Conclusions This study reveals that intercellular connections between chondrocytes contain GJs that play a key role in cell–cell communication and a metabolic function by exchange of nutrients including glucose and essential amino acids. A three-dimensional cellular network mediated through GJs might mediate metabolic and physiological homeostasis to maintain cartilage tissue.
- Chondrocytes
- Osteoarthritis
- Arthritis
Statistics from Altmetric.com
Introduction
Articular cartilage covers the ends of bones to facilitate the painless, low-friction movement of synovial joints. This tissue contains extracellular matrix (ECM) that is primarily composed of type II collagen and proteoglycans. Chondrocytes are highly specialised cells embedded in the ECM that provide cartilage with its remarkable mechanical properties. The cells occupy less than 10% of the total volume of articular cartilage but are responsible for the formation, maintenance and repair of the tissue throughout adult life.1 Unlike bone and other connective tissues, adult cartilage is an avascular, alymphatic tissue that is not innervated.2 ,3
Chondrocytes are isolated in small cavities known as lacunae. This characteristic has given rise to the notion that chondrocytes are not directly connected with each other and that they communicate with other chondrocytes primarily through the diffusion of substances in the matrix. However, proper cell-to-cell communication is essential for the organisation of a timely and uniform response to physical or biological stimuli or cell damage or simply to maintain tissue homeostasis. The lack of blood vessels and lymphatics and the relative isolation of the cells have been proposed to underlie the poor regenerative capacity of cartilage. This, however, is most likely not the case for articular cartilage, as ageing reduces the cell density and cartilage thickness, which allows the tissue to maintain its structure and function.4 ,5 However, the progressive degeneration of the articular cartilage matrix leads to osteoarthritis (OA), which is one of the most common causes of pain and disability in the Western world. OA is a degenerative joint disease characterised by the degeneration and subsequent loss of articular cartilage structure and function.6–9
We have recently shown that human articular chondrocytes express integral membrane proteins known as connexins (Cxs).10 Cxs form hemichannels that in turn form gap junctions (GJs) to directly connect the cytoplasm of two adjacent cells. GJs are responsible for the chemical and electrical coupling that occurs in neuronal synapses and the coordinated depolarisation of cardiac muscle.11 These channels enable the exchange of small hydrophilic molecules, including second messengers such as inositol 1,4,5-triphosphate (IP3), cAMP, glutamate and ATP,12–17 which serve a variety of functions. We have demonstrated that primary chondrocytes from human adults retain the capacity to communicate through voltage-gated GJ channels.10 The presence of functional GJs, together with other previously unpublished results obtained by our group using optical microscopy, led us to further investigate the morphology of articular chondrocytes in tissue.
Materials and methods
Cartilage collection and processing
Human knee and femoral head articular cartilage samples were obtained from adult donors after joint surgery after informed consent and Institutional Ethics Committee approval. Fresh cartilage explants from Sus scrofa were generously donated by the veterinary area of the INIBIC-Hospital Universitario A Coruña. In situ cartilage samples were frozen immediately in Cryomold Standard using Tissue-Tek O.C.T. compound and isopentane in liquid nitrogen and stored at −80°C. For the isolation and culture of primary chondrocytes, the fresh cartilage surface was rinsed with saline. A scalpel was used to cut parallel vertical sections, which were subsequently incubated with trypsin-EDTA solution (0.5 mg/mL) for 10 min at 37°C. After removing the trypsin solution, the cartilage slides were treated for 16–18 h with 2 mg/mL clostridial collagenase type IV (Gibco BRL, Invitrogen, Carlsbad, California, USA) in Dulbecco's modified eagle's medium (DMEM), supplemented with 5% foetal calf serum (FCS; Gibco, Invitrogen) to release the cartilage cells. The cells were then seeded onto 25 cm2 (250 000 cells), 75 cm2 (2 million cells) or 162 cm2 (3 million cells) flasks and incubated at 37°C in 5% CO2 and air in DMEM supplemented with 100 µg/mL Primocin (Invivo Gen Primocin) and 15% FCS.10 The cells were grown to ∼80–90% confluence. Chondrocytes were used for experiments during the third or fourth week of primary culture (see online supplementary figure S1).
Tissue processing and immunohistochemistry (IHC) assays
Cartilage sections were serially sectioned (4 µm) at −20°C in a Cryostat (Leica CM1510). The tissue sections were fixed with acetone for 10 min at 4°C, dried at room temperature (RT) and washed for 10 min with PBST (PBS with 0.1% Tween 20, pH 7.6). Before staining, endogenous peroxidase activity was inhibited by incubation with H2O2 and methanol for 10 min. The cells were then washed once with PBST, and the primary antibody was applied for 1 h at RT. After three washes with PBST for 10 min each, the sections were incubated with a peroxidase-labelled polymer conjugated to goat anti-mouse/rabbit Ig (Dako, Denmark) for 1 h. After three washes with PBST, peroxidase activity was developed using a freshly prepared substrate chromogen solution containing 3,3-diaminobenzidine tetrahydrochloride and H2O2 (Dako, Denmark). The sections were washed in distilled water, counterstained with Gill's haematoxylin, gradually dehydrated with graded alcohol and mounted in xylene with Depex (Serva, Germany). The cultured cells were seeded (25 000 or 250 000 cells) onto chamber slides and fixed with acetone for 10 min at 4°C, dried at RT and washed for 10 min with PBST before following the procedure described above. Negative controls (omitting the primary antibody) were performed to test the specificity of each antibody. Anti-Cx43 (610062) was supplied from BD Transduction Laboratories. Anti-Collagen II (MAB1330) was obtained from Millipore. Anti-Sox9 (ab76997) was purchased from Abcam and anti-CD31 (M0823) from Dako. Tissue sections were also counterstained with Gill's haematoxylin or with 4′,6-diamidino-2-phenylindole (DAPI). The slides were imaged using an Olympus BX61 microscope and a DP71 digital camera (Olympus). Image calibration was performed with AnalySISD V.5.0 software (Olympus Biosystems, Hamburg, Germany).
Scanning electron microscopy
Tissues frozen in the Tissue-Tek O.C.T. compound and stored at −80°C were cut into pieces with a scalpel, defrosted at RT by immersion in 0.2 M cacodylate buffer, pH 7.4, dehydrated in a graded series of ethanol, dried using the critical point method and coated with gold using a sputter procedure. Images were obtained at RT using a Jeol JEM 6400 Scanning Electron Microscope at 20 kV and Inca Energy V.200 software.
Co-immunoprecipitation
Immunoprecipitation (IP) experiments were performed with primary chondrocytes from human adults. Because lysis and solubilisation can disrupt protein–protein or protein–membrane interactions, to identify Cx43-interacting partners, the cells were cross-linked using a low concentration of formaldehyde (1%) for 15 min at RT.18 The cross-linking reaction was quenched for 10 min by adding 1 M glycine to a final concentration of 0.125 M. After cross-linking, the cells were collected, washed with PBS and frozen at −80°C. The cells were then resuspended in 1000 µL of IP lysis buffer containing protease inhibitors (Sigma) and 1 mM PMSF (50 mM Tris–HCl, pH 7.5, 5 mM EDTA, pH 8, NP-40 0.5% vol/vol, 1.0% vol/vol Triton-X and 150 mM NaCl). The lysates were precleared by incubation with 50 µL of protein G sepharose (50% slurry). The anti-CTD-Cx43 antibody (SC-20; Santa Cruz Biotechnology) was then added to the precleared lysates, followed by protein G sepharose beads (50 µL of 50% slurry), and the samples were incubated overnight at 4°C. To discard non-specific-binding proteins, Co-IP experiments were also performed in parallel without antibodies. The beads were washed four times with IP buffer. The bead pellets were suspended in loading buffer (10% SDS, 0.2 M Tris, pH 6.8, 50% glycerol, 0.1% (w/v) bromophenol blue (5×) containing 1.42 M 2-mercaptoethanol) and boiled for 10 min. The supernatants were collected and stored at −80°C. Silver stain analysis of the IPs and western blots was used to determine the specificity of the interactions. For SDS-PAGE gel electrophoresis, 5 µL of the denatured immunoprecipitated proteins were loaded and separated on 10% polyacrylamide gels. The gels were electroblotted onto polyvinylidene fluoride membranes (Millipore Co, Bedford, Massachusetts, USA). The membranes were blocked with 5% milk in PBS containing 0.1% Tween-20 (Sigma). After probing with antibodies, the membranes were developed using an ECL Western Blotting Detection reagent from GE Healthcare.
Identification of proteins using SDS-PAGE and nanoLC MALDI-TOF/TOF
Immunoprecipitated proteins were loaded and separated by SDS-PAGE on 10% acrylamide gels in a Protean mini-gel system (Bio-Rad, Hercules, California, USA). The gels were stained with Coomassie Brilliant Blue G25, and the resulting lanes were size-fractioned into three sections that were subsequently processed independently and subjected to in-gel digestion following a standard procedure.19 The peptide fractions were separated using reverse-phase chromatography in a nanoLC system (Tempo, Eksigent, Dublin, California, USA) and deposited onto a MALDI plate using an automatic spotter (SunCollect, Sunchrome, Friedrichsdorf, Germany). The mass spectrometry (MS) runs for each chromatogram were acquired and analysed in a MALDI-TOF/TOF instrument (4800 ABSciex, Framingham, Massachusetts, USA). The identification of peptides and proteins and the relative quantification of their abundances were performed using the Protein Pilot software V.3.0 (Applied Biosystems) with a Paragon Algorithm. The MS/MS data were searched against the UniProt/Swiss-Prot database of protein sequences (UniProt Swiss-Prot 2010_2012) using the following parameters: cysteine alkylation with IAA, urea denaturation, one missed cleavage allowed in trypsin digestion and focus on biological modifications. Only proteins with a threshold >95% confidence (>1.3 Unused Score) were considered for protein identification.
Image recording and data analysis
To determine the intercellular permeability of glucose and oligonucleotides, we employed previously published methods.20 ,21 Primary human chondrocytes were placed on an inverted microscope for probe injection. Micropipettes were pulled from capillary glass with a vertical pipette puller and backfilled with either 1 mM glucose or 0.5 mM of oligonucleotide in pipette solution as described previously.15 A 12-mer morpholino was used as previously reported.15 Both probes were injected into one cell of a monolayer of primary chondrocytes obtained from human adults, as previously reported.20 The data recording and analysis were performed as previously described.17 ,20
Electroporation in situ
The quantification of GJ intercellular transference was performed as previously described.22 ,23 Briefly, the system (InSitu Porator, Cell Projects Ltd) was used to introduce tracer dyes and other molecules into cells growing on a transparent electrode in a non-traumatic manner (cells located to the right of the electrode; see figure 5B), and GJ-based transfers are detected by diffusion of the dye to adjacent, non-electroporated cells (cells located to the left of the electrode). The cells were grown on glass slides, half of which was coated with electrically conductive, optically transparent, indium-tin oxide. A total of 400 µL of the fluorescent glucose tracer, 2-NBDG (1 mg/mL in HBSS without calcium), was applied to the cells and introduced through an electrical pulse (mild setting, 20 V). The electroporation solution was then aspirated, and the cells were incubated at 37°C for 4 min in DMEM supplemented with 10% dialysed FBS. The cells were then fixed with 4% formaldehyde in PBS for 2 min at RT. The migration of the dye through GJs to non-electroporated cells growing on the non-conductive area of the slide was observed under fluorescence illumination using an inverted fluorescence microscope (Inverted Research Microscope Eclipse Ti, Nikon) with imaging software (NIS-Element AR, V.3.0).
Metabolic capture assay
A layer culture system was used to study the transference of amino acids as previously described14 ,24–27 with some modifications. Briefly, ‘donor’ primary human chondrocytes were cultured in SILAC DMEM containing 10% dialysed FBS and supplemented with 73 mg/mL of [13C6]-L-lysine, 28 mg/mL of [13C6, 15N4]-L-arginine and 10× L-glutamine, designated ‘labelled’ medium. The same number of primary chondrocyte ‘receiver’ cells was cultured in non-labelled SILAC DMEM containing 73 mg/mL L-lysine and 28 mg/mL L-arginine, L-glutamine and 10% dialysed FBS. The cells were expanded in labelled or non-labelled medium for six doublings. The receivers were plated onto inverted inserts containing porous membranes suitable for cell culture (12 mm Transwell with a 3.0 µm Pore, Corning) or into each well of a six-well cluster plate as ‘control receivers’. After adhering to the membranes, the inserts containing receiver cells were turned right-side-up and plated into six-well plates. The donors and receivers are able to make contacts through cellular projections and to form GJs through the pores in the membrane, which measure 3 µm in diameter and are small enough to block cells greater than 10 µm in diameter from migrating to the other side of the membrane (cells incubated on the top of the membrane overnight did not migrate through the membrane) within the time frame of the assay (4 h after the donors were plated above the receivers). The cells were scraped with a sterile rubber policeman (Sigma-Aldrich), collected in PBS in 1.5 mL tubes (Eppendorf) and stored at −80°C. Each cell pellet was lysed in 112.5 μL of lysis buffer (10 mM Tris, pH 8.0, 10 mM EDTA, pH 8.0, 1% NP-40, 1 mM PMSF) for 20 min at 4°C, diluted with 634.5 μL of dilution buffer (10 mM Tris, pH 8.0, and 10 mM EDTA, pH 8.0), sequentially filtered through 50 and 3 kDa Centricon filters (KDa, Amicon) and frozen at −80°C. The fractions corresponding to molecules less than 3 kDa were dried in a SpeedVac (Savant SPD 121p, Thermo). Identification and quantification of free amino acids were performed using ESI*/LC/MS-Orbitrap. Prior to the LC analysis, the amino acids were derivatised using the EZ:faastTM kit according to the manufacturer's protocol (EZ:faast LC-MS for the free physiological amino acid kit; Phenomenex, USA). Calculations were performed using the data analysis portion of the software that controls the analytical system (Xcalibur V.2.0.7, Thermo Scientific, USA).
Statistical analysis
Data were analysed using GraphPad Prism software (V.5.00). Significant differences between sample groups were assessed using Student t test and Mann–Whitney test. Significant differences are represented as *p<0.05 and **p<0.01. Data are presented as mean±SEM.
Results
Articular chondrocytes have long, thin cytoplasmic arms extending from the cell body to connect with distant lacunae
Gill's haematoxylin-stained histological sections of cartilage from S scrofa revealed the presence of multiple projections that cross the matrix and specifically join cells located in different lacunae (see figure 1A and online supplementary figure S2). While these projections were primarily located in the superficial to middle zones, they were also observed in the deepest zones of the cartilage. IHC analysis of cartilage from human adults, performed with anti-Col2A and anti-Sox9 antibodies, suggested the presence of long cellular projections (figure 1B), ranging from 5 to 150 µm in length, between distant lacunae (see online supplementary figure S3). Cells with projections showed positive signals for both antibodies (figure 1B). A more detailed examination by confocal microscopy showed further evidence for the presence of long cytoplasmic extensions that project from the cell body and span several cell micrometres within the matrix to reach a distant lacuna (figure 1C).

Optical images demonstrate the presence of cellular connections between chondrocytes in articular cartilage. (A) Immunohistochemistry (IHC) coupled with Gill's haematoxylin counterstaining using articular cartilage from Sus scrofa (3 months old) and an antibody for the endothelial/haematopoietic marker CD31 confirmed that the cells found in individual lacunae and containing cellular connections correspond to chondrocytes (see online supplementary figure S2). Cartilage sections were analysed from the superficial zone, located next to the synovial fluid, to the deepest region next to the subchondral bone. (B) IHC analysis of cryosections of cartilage obtained from human adults stained with a monoclonal anti-Col2A antibody or an anti-Sox9 antibody. Note that no Gill's haematoxylin counterstaining was performed before IHC experiments. (C) Confocal microscopy confirmed that cell projections and cell bodies are embedded in the extracellular matrix and that cell projections and cell bodies coincide in the same point of plane. Transmitted light images were obtained using a Nikon Ti Eclipse Confocal Microscope and the Confocal A1R Nikon imaging system. The analysis was performed using NIS Elements AR V.3.2 imaging software. For 3D imaging, sequence of Z-stack images (each 0.53 µm of depth) were captured in different Z-axis (depth of focus). The images are shown in X/Y axis and XZ/YZ (Z-stack images) axis. The green dashed line shows the cell body and cell projection detected in the same plane of section.
The structure of the cartilage does not favour the identification of cell elements, projections or other cellular details that cross the dense ECM by microscopy techniques. Cellular extensions between cells in the same plane of sectioning were only detected in less than 1% of the analysed samples. The results obtained in figure 1A were obtained using young cartilage from S scrofa, which contains higher number of chondrocytes than cartilage from human adults.
Scanning electron microscopy (SEM) of S scrofa and human cartilage samples confirmed that the projections observed by optical microscopy (figure 1) are indeed cytoplasmic extensions of the cell (figure 2). Chondrocytes within cartilage contain at least two long cytoplasmic extensions per chondrocyte that spread thinly (∼200 nm in width) along the ECM and were found to physically connect cells located in different lacunae (figure 2).
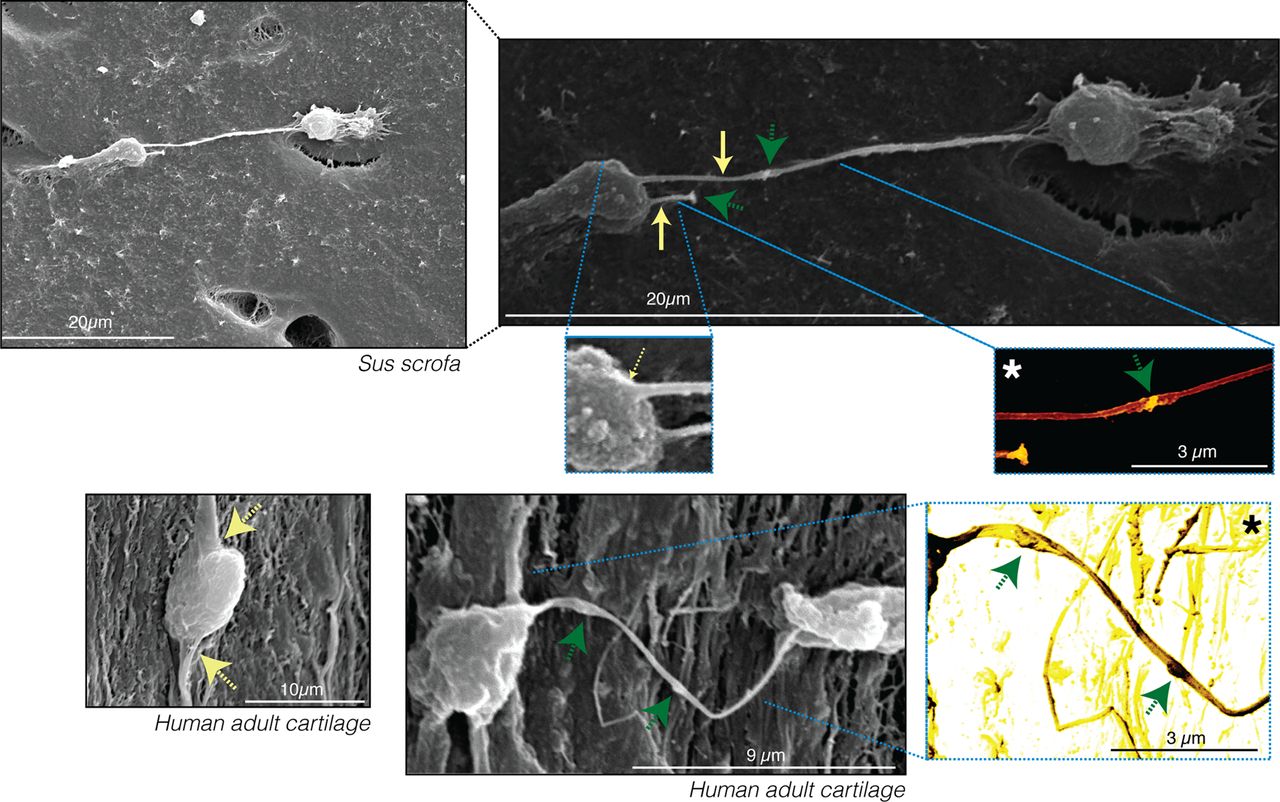
Scanning electron microscopy images validate the presence of long, thin cytoplasmic arms extending from the cell body. Long cytoplasmic arms were observed in chondrocytes from Sus scrofa and human adults. The yellow arrows indicate cytoplasmic projections. Original images show different characteristic intensities or grey levels. The cells appear bright grey, whereas the matrix components are darker grey. The asterisk indicates images that were processed to improve the contrast and provide colour to the original greyscale image. Homogeneous colourisation was performed for the entire image without selecting any areas to ensure that colours fairly represent the differences in the intensity of the original images. The green arrows indicate the presence of vesicles or other components within the projection.
Cx43 is located along chondrocyte membranes, including cellular projections
As predicted, IHC analysis of primary human chondrocytes revealed high levels of Cx43 between the cytoplasm of two adjacent cells, confirming the presence of GJ channels between the contacting cells (figure 3A, blue arrows). Interestingly, Cx43 was also detected along the thinner extensions of cultured human chondrocytes (figure 3B, red arrows). In tissue samples, Cx43-positive spots were distributed throughout the cell, particularly at the edges of the lacuna (figure 3C). We also detected positive staining within the matrix outside the lacuna (figure 3C, red arrows), suggesting the presence of GJs in the cellular projections.

Interaction of Cx43 with microtubules and actin filaments. (A) Immunohistochemistry (IHC) with an anti-Cx43 antibody coupled with Gill's haematoxylin counterstaining on primary human chondrocytes. Negative control is shown on the right. The nucleus (dark purple) can be differentiated from the cytoplasm (clear purple). Cx43 was primarily localised in the cytoplasm, at the edge of the cytoplasm (membrane localisation), between the cytoplasm of two adjacent cells (blue arrows) and (B) in the thin extensions (red arrows). (C) IHC with anti-Cx43 on articular cartilage from adult humans. Positive signals for Cx43 were enriched on the cell edges (first and last image) and in the middle of the matrix (red arrows), suggesting the presence of thin extensions in the latter. (D) Immunoprecipitation (IP) experiments combined with the identification of interacting proteins through mass spectrometry analysis. Whole cell extract (WCE) and proteins immunoprecipitated with an anti-CTD-Cx43 antibody were analysed by silver staining and western blotting. Remnants of the IgG-heavy chain (53 kDa) from the antibody used for IP are visible but distinguishable from the Cx43 signal (43 kDa). The proteins listed in table 1 were identified from ten IP experiments using primary chondrocytes from ten different donors.
Interactions between Cx43 and cytoskeletal proteins implicated in Cx trafficking were investigated through co-immunoprecipitation using cultured human chondrocytes and an antibody that specifically recognises the C-terminal end of Cx43, followed by nano-LC-MS/MS analysis. The MS data analysis indicated that the C-terminal tail of Cx43 interacts with several cytoskeletal proteins, including α-tubulin and β-tubulin, actin, vimentin and vinculin (figure 3D and table 1). These results are consistent with chondrocytes forming GJs and communicating through cellular extensions.
Cx43-interacting proteins identified by nanoLC-MS/MS
Neighbouring chondrocytes exchange solutes, including oligonucleotides
An analysis of fluorescent DAPI counterstaining of cartilage sections from S scrofa revealed positive fluorescence in cellular projections (figure 4A), suggesting functional coupling among tissue chondrocytes. The images shown in figure 4A were overexposed; however, DAPI fluorescence was specifically observed in cellular projections, not in the matrix, indicating that these fluorescent signals were not due to overexposure of the image.

Direct exchange of small oligonucleotides. (A) 4′,6-Diamidino-2-phenylindole (DAPI) staining of cartilage from Sus scrofa. Optical and fluorescent images reveal fluorescent cellular projections. The images were overexposed. Original and processed images are shown as indicated. (B) Flux of a 12-mer oligo in human chondrocyte cell pairs. Upper panel: a pipette containing the oligo was attached to the cell on the right in a whole-cell configuration. Epifluorescent micrographs were captured at 2, 4, 6, 8, 10 and 12 min after dye injection into the cell on the right. The results show progressively increased fluorescence intensity in the recipient cell. Lower panel: plots of fluorescence intensity versus time for the injected cell (filled circles) and the recipient cell (empty circles). The data are presented as mean±SEM.
To confirm whether adult human chondrocytes can transfer siRNA-sized oligonucleotides through GJs, as suggested by the images obtained in figure 4A, fluorescently labelled oligonucleotides (morpholinos), 12 nucleotides in length (12-mer oligo), were synthesised and introduced into one cell of a pair using a patch pipette (figure 4B). The results demonstrated that the 12-mer oligo was transferred between primary chondrocytes within 12 min after injection (figure 4B).
Direct transfer of glucose and amino acids between neighbouring chondrocytes
The capacity of chondrocytes to transfer glucose through GJs was studied by loading glucose into individual human chondrocytes and subsequently confirmed through in situ electroporation (figure 5). A solution containing a fluorescent glucose tracer (2-NBDG) was introduced into one cell of a pair (donor cell) through a patch pipette (figure 5A). Intercellular transfer of glucose was observed between adjacent chondrocytes, resulting in homogeneous distribution of the label within 22–35 min (figure 5A).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Direct transfer of nutrients between chondrocytes. (A) Glucose flux in contacting human chondrocytes. A pipette containing fluorescently labelled glucose was attached to a cell (yellow arrow). Glucose transference was monitored, and images were captured at 22 and 35 min after dye injection. (B) In situ electroporation on a partly conductive slide for the measurement of intercellular junctional transfer of glucose. Primary human chondrocytes or human lung carcinoma cells (line A549) were electroporated in the presence of 1 mg/mL fluorescently labelled glucose. After washing the unincorporated glucose, the cells were photographed under phase contrast or fluorescence illumination. The red lines define the conductive (left) and non-conductive sides (right). Gap junction (GJ) intercellular communication was severely reduced when cells were pretreated with 250 µM of GAP27 for 1 h. Original magnification ×10. (C) Quantification of glucose transfer through GJs. The graph represents the number of contacted cells into which the dye was transferred per electroporated border cell (score). The data are presented as mean±SEM. n=6; **p<0.01; Mann–Whitney test: untreated versus GAP27, and chondrocytes versus A549. (D) Transwell-layered culture system used to study the transfer of amino acids through cellular projections and GJ channels. IHC with an anti-Cx43 antibody on transwell membranes containing articular chondrocytes from adult humans. The green arrow indicates a cellular projection within the pore. The donor cells were previously cultured in labelled medium containing 73 mg/mL [13C6]-L-lysine and 28 mg/mL of [13C6, 15N4]-L-arginine and plated on top of membranes that separated the cells from the receiver cells cultured in non-labelled medium. The data are presented as mean±SEM. n=4 for L-arginine, and n=4 for L-lysine. **p=0.0087, *p=0.0286; Mann–Whitney test: receivers versus controls.
In situ electroporation on a partly conductive slide confirmed that the direct transfer of glucose between contacting cells occurred through GJs (figure 5B). Primary chondrocytes displayed a gradient of fluorescently labelled glucose following electroporation in the in situ chamber (figure 5B). Quantification of intercellular communication revealed extensive transfer (8–20 cells deep, score 7.39) of glucose between cells (figure 5C). The graph shows the number (score) of contacted cells into which the dye was transferred per electroporated border cell. No transference was detected when glucose was electroporated into A549 cells, which have no GJs (0–1 cell deep, score 0.16). In addition, inhibition of GJ intercellular communication using the Cx43 mimetic peptide blocker GAP27 (250 µM for 1 h) significantly reduced the transfer of glucose between chondrocytes (1–4 cells deep, score 1.61) (figure 5B, C).
To study the transfer of endogenous amino acids through GJs, we employed stable isotope labelling of amino acids in cell culture (SILAC) in a layered culture system containing a porous membrane (figure 5D). IHC analysis of transwell membranes containing human articular chondrocytes stained with an anti-Cx43 antibody revealed the presence of cellular projections through the pores 4 h after the cells were plated (figure 5D). We included a layer of ‘control receiver’ cells plated 1 mm below the membrane to prevent direct cell contact or GJ formation with donors (figure 5D). MS analysis revealed the presence of L-lysine (13C6) and L-arginine (13C6-15N4) in the receivers but not the controls, confirming the intercellular transfer of these amino acids (figure 5D).
Discussion
In the present study, we demonstrate for the first time that chondrocytes in articular cartilage are physically connected through a cellular network capable of mediating metabolic coupling and the transfer of small siRNA-sized or miRNA-sized RNA molecules, as well as glucose and essential amino acids. SEM analysis revealed that chondrocytes embedded in the cartilage matrix contain at least two long cytoplasmic arms that spread along the matrix to reach a distant cell located in a different lacuna (figure 2). Long thin extensions of the cytoplasm were also observed in cultured chondrocytes (figure 3A,B).
A number of studies have elucidated structural characteristics of articular chondrocytes. The presence of small cellular extensions was observed in the superficial layer of cartilage from one patient with OA.28 In addition, cell processes 8 µm in length were observed in the middle zone29 and identified by others as potential interconnecting tracks running through the ECM.30 Although some reports suggested the presence of cytoplasmic projections or cell remnants in the superficial and middle zones, no previous studies have directly observed intercellular connections between chondrocytes in the cartilage matrix.
The existence of intercellular connections relies on the establishment of cell–cell contacts. Recently reported results from our group revealed that cell-to-cell communication between adult human chondrocytes can occur through GJ channels.10 In fact, it was previously shown that animal chondrocytes express the GJ protein Cx43 and form functional GJ channels in dense monolayer and micromass culture.31–38 However, these reports fail to explain how highly specialised cells such as articular chondrocytes that exist as individual cells that are not in physical contact with one another express Cx43 and form GJ channels. It has been assumed that GJs only occur in clusters of chondrocytes. Results presented herein demonstrate that distant chondrocytes in mature articular cartilage are in physical contact with one another and that cell-to-cell communication through GJ channels allows for the exchange of various different molecules.
DAPI is a cationic compound that is nearly impermeable to the plasma membranes of live cells; therefore, it is typically used for staining nuclei of fixed and dead cells. However, the transfer of DAPI through GJ channels can be used to study functional coupling between living cells.39–41 The positive fluorescence observed in the cellular projections (figure 4A) of chondrocytes in in situ cartilage suggests that chondrocytes in vivo within cartilage are functionally coupled. The results presented in figure 4B further indicate that these cells are capable of transferring small molecules of RNA. Endogenous and exogenous siRNA or mature miRNA can directly alter gene expression. Several reports have revealed the potential importance of miRNA in maintaining cartilage integrity and homeostasis.42–44 In fact, the major chondrogenic transcription factor Sox9 positively regulates the COL2A1 gene in human chondrocytes by a miR-675-dependent mechanism.45 Through the transfer of small RNAs, a small group of cells or a single chondrocyte has the potential to affect gene expression in a larger group of cells or throughout the cartilage.
GJ communication is required for physiological processes such as growth and metabolic coordination.46 Metabolic coupling has special relevance in the case of avascular tissues such as the cornea or lens. In these tissues, three-dimensional cellular networks and GJs are essential for the maintenance of metabolic and physiological homeostasis of the tissue, which are in turn essential for the maintenance of corneal transparency. Several congenital mutations of lens fibre cell Cxs cause various types of cataracts by affecting the conductance and/or gating of GJ channels or altering protein trafficking or channel assembly.47 We recently reported that altered Cx protein levels and subcellular localisation are most likely associated with the development of OA.10
Cxs assemble into functional hexameric connexons in the endoplasmic reticulum membrane and pass through the Golgi apparatus to reach the plasma membrane by interacting with tubulin and actin filaments.48 ,49 The results shown in figure 3 and table 1 confirm that Cx43 interacts with the microtubules and actin filaments implicated in the shuttling of connexins throughout the cytoplasm of articular chondrocytes, to be later inserted into any site on the membrane, including cellular projections. We also detected other Cx43-interacting proteins, including erzin and vinculin, which can affect GJ assembly.49
Sinovial and bone cells express Cx proteins and form functional GJ channels.37 Osteocytes contain multiple cellular projections and processes that, together with GJs, comprise a complex intercellular communication network implicated in matrix deposition, mechanotransduction and architecture in bone.37 Cx43-expressing cells transfer molecules to other Cx43-expressing cells,15 and the deepest zone of cartilage is contiguous with the subchondral bone. Future studies should be performed to determine if the cells that form the three main tissues of the joint are capable of intercellular communication.
The establishment of cell–cell contacts that allow GJ intercellular communication is essential for the normal function of vascular and avascular tissues. Cartilage health is critical for the maintenance of mobility and quality of life in humans. The results presented herein represent a crucial contribution to our understanding of normal cartilage function and might lead to the identification of biological mechanisms underlying cartilage diseases, including the eventual degeneration of the matrix, which occurs in patients with OA.
In summary, the data reported here demonstrate that chondrocytes in articular cartilage form a three-dimensional cellular network mediated by GJs and provide strong evidence that such intercellular connections may be involved in the synchronised regulation of the metabolic and physiological cartilage homeostasis.
Acknowledgments
The authors would like to thank Lourdes Sanjurjo and Dolores Salinas Bujan for generously providing cartilage samples after surgery; Maria Jose Sanchez Dopico and Dr Tamara Hermida for isolating articular chondrocytes from cartilage for cell culture; Dr Catalina Sueiro Lopez and Dr Ada Castro-Couceiro (SAI, UDC) for helpful suggestions and sample preparation for SEM; Dr Veronica Fernandez-Villarenaga Martin and Dr Gerardo Fernandez Martinez (SAI, UDC) for suggestions, sample preparation and data analysis (LTQ-Orbitrap); Dr Moises Blanco for the statistical advise; Dr Leda Raptis for helpful discussions and suggestions; the Translational Cancer Research Group of INIBIC for the A549 cell line; members of the proteomics group (INIBIC) for helpful technical suggestions; Christian Steinhäuser research group for advise and electrophysiology experiments on tissue; and Estefania Cives for administrative assistance.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figures
Footnotes
Handling editor Tore K Kvien
MDM, RG-F and PC-F contributed equally.
Contributors All authors were involved in drafting or critically revising the manuscript for important intellectual content, and all authors approved the final version for publication. FJB and MDM have full access to all of the data in the study and take responsibility for the integrity and accuracy of the data presented. MDM and FJB are responsible for study conception and project planning.MDM, PC-F, RG-F, PF-P, PF-F, NG, VV, PRB, GSG and FJB are responsible for acquisition of data.MDM, VV, PRB, GSG and FJB are responsible for data analysis and interpretation.
Funding This work was supported in part through funding from the Fondo Investigación Sanitaria, Madrid, Spain (CIBER-CB06/01/0040; PI12/00329; RETIC-RIER-RD12/0009/0018; and Proteo-Red/ISCIII) (to FJB); Ministerio Ciencia e Innovación PLE2009-0144, FEDER (European Community) (to FJB); and the National Institutes of Health grants R01 GM088181 (to VV) and PRB-NIH RO1 GM088180 (to PRB). This work was also supported through funding from a CIBBER-BNN fellowship, a predoctoral fellowship from Xunta de Galicia to RG-F and a grant from Fondo Investigación Sanitaria-Spain (CA09/00458) to PF-P. MDM is an Isidro Parga Pondal researcher (Xunta de Galicia).
Competing interests None.
Patient consent Obtained.
Ethics approval The study was conducted with the approval of the local ethics committee in Galicia, Spain.
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Correction