Article Text

Abstract
Objectives Blockade of transient receptor potential vanilloid 1 (TRPV1) with systemic antagonists attenuates osteoarthritis (OA) pain behaviour in rat models, but on-target-mediated hyperthermia has halted clinical trials. The present study investigated the potential for targeting TRPV1 receptors within the OA joint in order to produce analgesia.
Methods The presence of TRPV1 receptors in human synovium was detected using western blotting and immunohistochemistry. In a rat model of OA, joint levels of an endogenous ligand for TRPV1, 12-hydroxy-eicosatetraenoic acid (12-HETE), were quantified using liquid chromatography-tandem mass spectrometry (LC-MS/MS). Effects of peripheral administration of the TRPV1 receptor antagonist JNJ-17203212 on afferent fibre activity, pain behaviour and core body temperature were investigated. Effects of a spinal administration of JNJ-17203212 on dorsal horn neuronal responses were studied.
Results We demonstrate increased TRPV1 immunoreactivity in human OA synovium, confirming the diseased joint as a potential therapeutic target for TRPV1-mediated analgesia. In a model of OA pain, we report increased joint levels of 12-HETE, and the sensitisation of joint afferent neurones to mechanical stimulation of the knee. Local administration of JNJ-17203212 reversed this sensitisation of joint afferents and inhibited pain behaviour (weight-bearing asymmetry), to a comparable extent as systemic JNJ-17203212, in this model of OA pain, but did not alter core body temperature. There was no evidence for increased TRPV1 function in the spinal cord in this model of OA pain.
Conclusions Our data provide a clinical and mechanistic rationale for the future investigation of the therapeutic benefits of intra-articular administration of TRPV1 antagonists for the treatment of OA pain.
- Osteoarthritis
- Synovitis
- Knee Osteoarthritis
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
Osteoarthritis (OA), a degenerative disease of synovial joints, is a major cause of pain and physical disability.1 ,2 OA pain arises partly from altered sensory processing in the joint,3 ,4 as indicated by analgesic effects of intra-articular local anaesthetics, and lowered peripatellar pressure pain thresholds.5 ,6 The identification of substrates underpinning the sensitisation of sensory afferents innervating the OA joint may provide new targets for treatments which prevent, or delay, the progression of OA pain.
The pronociceptive non-selective cation channel transient receptor potential vanilloid 1 (TRPV1) plays important roles in the detection of noxious stimuli and inflammatory hyperalgesia.7 TRPV1 has been implicated in OA pain, both in animal models8–11 and by the finding that TRPV1 genetic variants are associated with the risk of symptomatic knee OA in humans.12 TRPV1 is enriched in small diameter cell bodies of nociceptive nerve fibres that innervate the articular capsule of the joint and is upregulated in the sensory afferent fibres innervating the OA joint.13
Clinical trials of oral TRPV1 antagonists have been limited by on-target-induced hyperthermia,14 which is also apparent in rodents15 ,16 and attributed to effects on the gastrointestinal system.17–19 Identification of the contribution of local knee joint, versus central, sites of action to the analgesic effects of TRPV1 antagonists in OA may provide novel approaches by which TRPV1 targeted analgesia can be achieved in the absence of side effects. Here we quantify levels of TRPV1 protein in human OA synovium, inflammatory rheumatoid arthritis (RA) synovium and postmortem (PM) controls and test the hypothesis that joint TRPV1 contributes to altered sensory inputs from the OA joint. Finally, we determine whether blockade of knee joint TRPV1 attenuates OA pain responses in the absence of hyperthermic side effects.
Methods
Subjects
We used synovial tissue from people undergoing total knee joint replacement (TKR) for OA (n=27) or RA (n=8) and PM knees from seven people without history of knee pain and with no macroscopic evidence of arthritis. Median age of participants was 68 (IQR 64–77) years). Human tissue collection followed informed consent from the donor or next of kin according to protocols approved by the North Nottinghamshire local research ethics committee (NNHA/420, NNHA/544 and NNHA/673).
Animals
Experiments were conducted on male Sprague–Dawley rats (160–190 g; Charles River, UK) in accordance with the Animal (Scientific Procedures) Act 1986 and ARRIVE guidelines. A total of 176 rats were used.
Assessment of hyperthermia
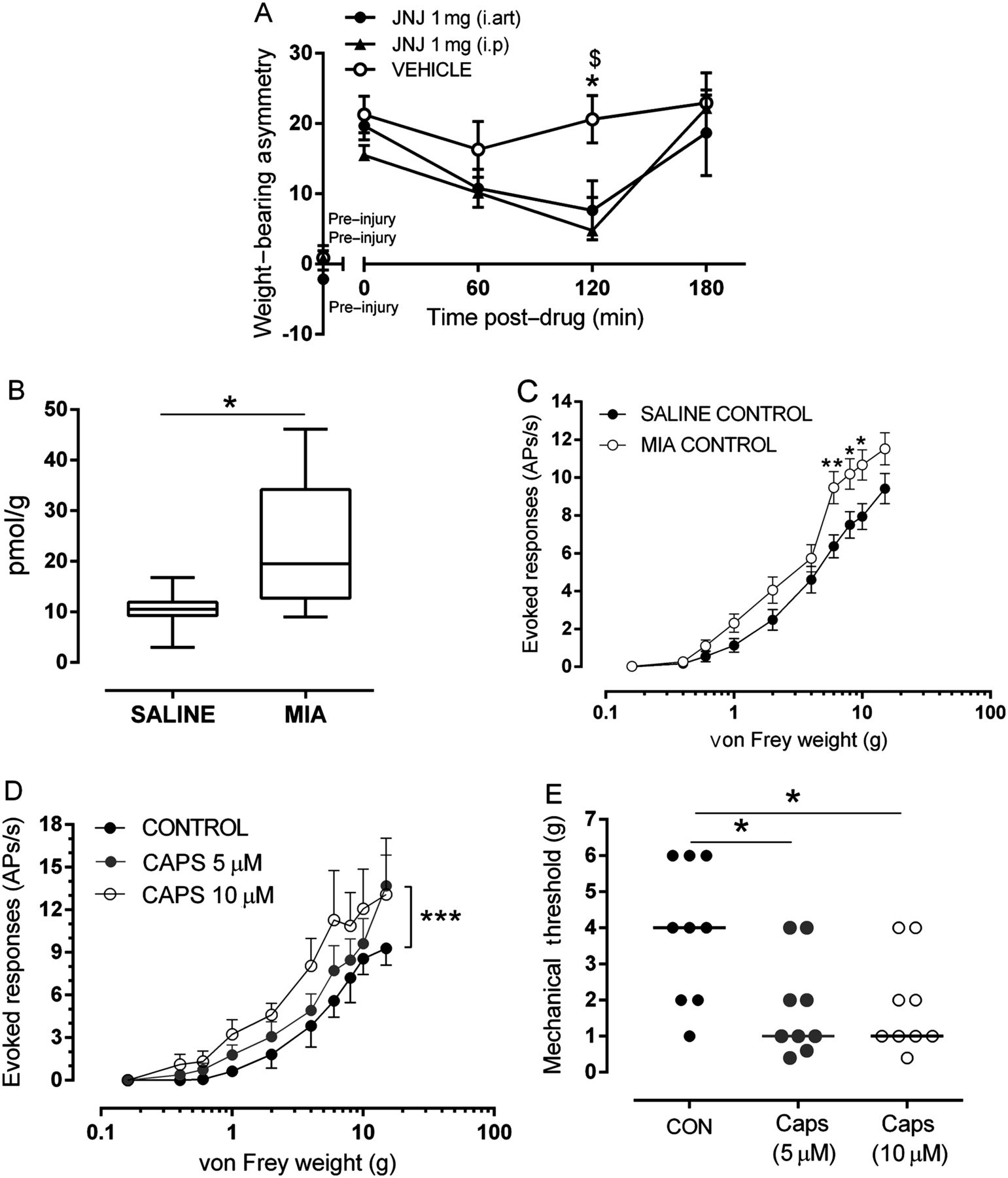
Under isoflurane anaesthesia, rats received an intra-articular injection of the TRPV1 antagonist JNJ-17203212 (1 mg/50 µL, n=6) or vehicle (3% Tween 80 in saline, 0.5% ethanol (EtOH), n=6). As a positive control,20 a separate cohort of rats received oral JNJ-17203212 (10.6 mg/1.05 mL, n=6) or vehicle (polyethylene glycol 400, n=6). Rectal temperature was measured up to 4 h post-drug administration.
MIA-induced OA and pain assessment
The mono-iodoacetate (MIA) model, which mimics common features of human OA joint pathology and is associated with robust pain behaviour21 ,22 was generated as previously described (see online supplemental methods).22 Pain responses were assessed as previously described,22 weight-bearing is presented as (weight on contralateral limb—weight on ipsilateral limb)/total weight on limbs×100) and weight-bearing asymmetry is indicative of hyperalgesia.23 Changes in hind paw mechanical withdrawal thresholds were assessed using von Frey (vF) monofilaments (Linton Instrumentation, bending forces 1–26g) as previously described.22 Rats were tested on days 0 and 3–28 post-injection.
Effects of intra-articular and systemic (intra-peritoneal (i.p)) injection of the TRPV1 antagonist JNJ-17203212 (1 mg/50 μL and 1 mg/300 μL, respectively, based on published studies20; Tocris, UK; n=11/7 rats), or vehicle (3%Tween 80 in saline, 0.5% EtOH; n=9/6 rats), on weight-bearing and hind paw withdrawal thresholds were assessed in MIA-treated rats at 14 days post-model induction; a time point associated with joint afferent sensitisation.24
Measurement of 12-HETE by LC-MS/MS analysis
Rats were humanely killed and knee joints, dorsal root ganglia (DRG) and spinal cord were rapidly dissected and stored at −80°C. The liquid chromatography-tandem mass spectrometry (LC-MS/MS) method employed is based on,25 see online supplemental methods.
TaqMan quantitative real-time PCR (RT-PCR)
Rats were humanely killed and knee joint synovium was rapidly dissected and embedded in optimal cutting temperature (OCT) compound. The real-time PCR (RT-PCR) method employed was based on,22 see online supplemental methods.
Joint afferent recordings
At 14 days following injection of MIA (n=30) or saline (n=37) rats were anaesthetised (sodium pentobarbital) and prepared for joint afferent recordings.24 Action potentials were recorded from two to three identifiable ipsilateral saphenous nerve filaments (Spike2 software, Cambridge Electronic Design (CED), Cambridge, UK) and firing rates of the most reliable single fibre were quantified.24 Mechanically evoked responses (the mean frequency of firing) of fibres following application of vFs (0.16–15g/5 s each) to subcutaneous structures over and around the knee joint were recorded. Thresholds and conduction velocities were calculated as described.24 Conduction velocities ranged from 0.31 to 13.4 m/s encompassing C-fibres and Aδ-fibres (n=67; one fibre studied per rat).
The TRPV1 antagonist JNJ-17203212 (0.075, 0.15 mg/100 μL), the TRPV1 agonist capsaicin (5, 10 μM/100 μL) or vehicle (100 μL 2.5, 5% ethanol/Tween 80 in saline) was administered by close intra-arterial administration via a right femoral artery cannula with the tip located at the bifurcation of the descending aorta24 and mechanically evoked responses were assessed every 5 min for 60 min/dose.
Spinal neuronal recordings
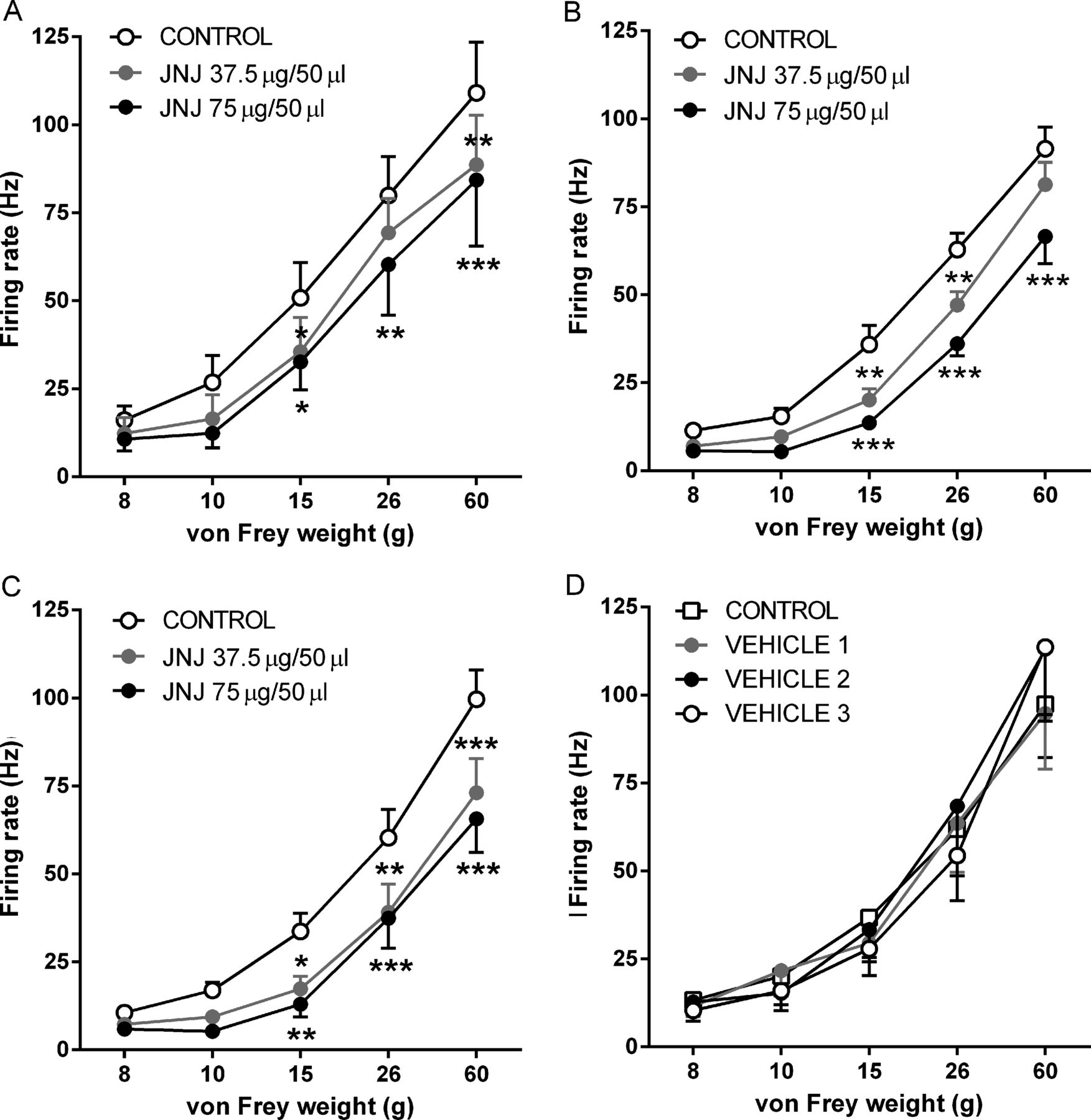
Rats were prepared as previously described.22 A sharp glass-coated tungsten microelectrode was inserted into the dorsal horn of the spinal cord. Wide dynamic range (WDR) neurones with a receptive field over the ipsilateral hind paw were recorded (depth 500–1000 µm from the cord surface). vF monofilaments (8, 10, 15, 26 and 60 g) were applied to the centre of the receptive field and evoked responses recorded. Effects of topical spinal administration of the TRPV1 antagonist JNJ-17203212 (18.75, 37.5 and 75 µg/50 µL) at 50 min intervals (n=7–9) or three equivalent doses of 50 µL vehicle (n=7) on evoked responses were recorded.
Immunohistochemistry
Immunohistochemistry26 with an affinity-purified rabbit polyclonal antibody directed against human TRPV1 (SC-20813, Santa Cruz Biotechnology Inc.) was conducted on synovial sections from patients with OA (n=9; three men/six women), RA (n=8; two men/six women) and PM controls (n=7; six men/one woman). TRPV1 and macrophage (CD68) immunoreactivities in synovial sections from arthritic human (n=6) were colocalised by double immunofluorescence (see online supplemental methods). TRPV1 fractional area was defined as the percentage of synovial tissue section area (within 200 μm of the synovial surface) occupied by TRPV1 immunoreactivity. Quantification was undertaken in a blinded fashion with computer-assisted image analysis as described previously.26 Rat spinal cord sections from MIA- and saline-treated rats (n=5/group) (see online supplemental methods) were incubated with a polyclonal guinea pig anti-TRPV1 antibody (1 : 500, Neuromics, Edina, Minnesota, USA catalogue number GP14100) and then with Alexa 568-conjugated goat anti-guinea pig secondary antibody (1:300, Molecular Probes). TRPV1 immunostaining was visualised with a Leica DMRB/DM4000 B fluorescence microscope and images were acquired using Openlab software (PerkinElmer). Quantification with Velocity V.5.5 software (Perkin Elmer, UK) used a minimum of five sections per rat. Negative controls (primary antibodies omitted) were included to confirm specificity of detected immunoreactivities.
Western blotting
Membrane-enriched fractions of OA synovial samples (n=18 patients) were prepared. Samples were homogenised in 2 mL NaCl-EDTA-Tris (NET) buffer (150 mM NaCl, 3 mM EDTA, 50 mM Tris, pH 7.4) containing 50 µL protease inhibitor cocktail (comprised of 4-(2-aminoethyl)benzenesulfonyl fluoride, pepstatin A, E-64, bestatin, leupeptin and aprotinin) and ultracentrifuged (1000×g, 10 min, 4°C). Membrane-enriched fractions were pelleted from the supernatant by ultracentrifugation (100 000×g, 30 min, 4°C) and resuspended in 200 µL NET buffer plus 0.5% triton x-100. One hundred microlitres of samples were separated on a 7.5% polyacrylamide gel and transferred to polyvinylidene difluoride membrane and exposed to anti-human TRPV1 for 24 h at 4°C and then a goat anti-rabbit horseradish peroxidase conjugate secondary antibody (PI-1000, Vector Laboratories, Cambridge, UK). The enhanced chemiluminescent (ECL) chemiluminescence kit was used to detect immunoreactivity by exposure to autorads.
Data and statistical analysis
Analysis was performed using Prism V.5 (Graphpad Software, San Diego, California, USA) and normality assessed (D'Agostino & Pearson normality test) prior to statistical analysis. t Tests compared two groups of data and analysis of variance (ANOVA) with Bonferroni's post hoc test compared more than two groups when normality was achieved. Non-parametric tests, Mann-Whitney (two groups) and Kruskal–Wallis with Dunn's post hoc test (more than two groups) were used when normality was not obtained. Behavioural, electrophysiological and spinal cord immunohistochemistry data are presented as mean±SEM.
Results
Increased expression of TRPV1 in human synovium
TRPV1 immunoreactivity was present in OA (figure 1A), RA and PM synovium and was predominantly localised to mononuclear cells within the synovial lining region, but was also within the deeper layers of the synovium where macrophages were abundant. The majority of TRPV1 immunoreactivity was localised to CD68-positive macrophages (figure 1A,B). TRPV1 fractional areas differed between disease groups (χ2=12.0, p=0.003, figure 1C). TRPV1 fractional areas were lowest in PM synovium (median 0.0%, IQR 0.0–3.3%) compared with patients with OA (median 7.3%, IQR 5.9–9.2%, Z=−2.9, p=0.004) or RA (median 5.2%, IQR 4.5–7.5%, Z=−2.6, p=0.01, figure 1C). The absence of staining in the negative control confirmed specificity of detected immunoreactivity. Membrane-enriched synovial samples from 12 (67%) of 18 OA patients displayed detectable TRPV1 immunoreactivity in the 100 kDa region (figure 1D), consistent with the reported molecular weight for TRPV1.27

Transient receptor potential vanilloid 1 (TRPV1) and CD68 immunoreactivities in human synovium. TRPV1 (A) and CD68 (B) immunoreactivities in synovium from a patient with osteoarthritis (OA). Immunoreactivities both for TRPV1 (green) and for CD68 (red) colocalise to mononuclear cells within the synovial lining and sublining regions (arrows). Occasional CD68 immunoreactive cells do not display TRPV1 immunoreactivity (arrowheads). L: surface of synovial lining. Double fluorescence immunohistochemistry. Scale bar=50 μm. (C) Box and whisker plots showing increased abundance of TRPV1-immunoreactive cells in OA compared with non-arthritic postmortem (PM) control synovium. **p=0.003. (D) Western blot analysis of TRPV1 in membrane-enriched fractions of synovial from five patients with OA. TRPV1 immunoreactivity was detected at approximately 100 kDa in lanes 1, 3, 4 and 5.
Intra-articular administration of a TRPV1 antagonist inhibits pain behaviour in the MIA model of OA pain
The next series of experiments used an experimental model of OA pain to investigate the potential contribution of joint TRPV1 to OA pain. Following intra-articular injection of MIA, rats exhibited a significant weight-bearing asymmetry and a significant decrease in hindlimb withdrawal thresholds compared with saline-treated rats (see online supplementary figure S1). Intra-articular injection of the TRPV1 antagonist JNJ-17203212 significantly attenuated MIA-induced weight-bearing asymmetry (figure 2A) compared with MIA-treated rats receiving vehicle and to a similar degree as that seen following systemic JNJ-17203212 (figure 2A). Unlike systemic JNJ-17203212, intra-articular JNJ-17203212 did not significantly alter distal allodynia (see online supplementary figure S2) in MIA-treated rats. Intra-articular injection of JNJ-17203212 did not alter core body temperature (baseline=37.08±0.35°C; 0.5 h=36.98±0.40°C, 2 h=37.42±0.11°C) compared with vehicle-treated rats (baseline=37.37±0.10°C; 0.5 h=36.90±1.2°C, 2 h=37.30±0.12°C). Note systemic administration of JNJ-1720312 significantly elevated core body temperature as previously described (see online supplementary data).

Endogenous transient receptor potential vanilloid 1 (TRPV1) activation at the level of the joint contributes to pain. (A) Intra-articular administration of JNJ-17203212 significantly attenuated weight-bearing asymmetry compared with intra-articular vehicle (3% Tween 80, 0.5% EtOH in saline) in rats 14 days post-mono-iodoacetate (MIA) injection (n=11/9 rats) and to a similar degree as systemic (intra-peritoneal (i.p)) JNJ-17203212. Vehicle effects following intra-articular and i.p administration were not significantly different; for clarity, intra-articular vehicle is presented. Data are presented as mean±SEM and analysis used a two-way analysis of variance (ANOVA) with a Bonferroni's post hoc test (weight-bearing) or Mann–Whitney test (allodynia). *p<0.05 between i.art JNJ-17203212 and i.art vehicle. $p<0.05 between i.p JNJ-17203212 and i.art vehicle. (B) Levels of the endogenous TRPV1 ligand 12-hydroxy-eicosatetraenoic acid were significantly greater in joint tissue from MIA-treated rats compared with saline-treated rats (p<0.05, Mann–Whitney; n=8 rats per group). (C) Stimulus–response curves of joint afferents to mechanical von Frey monofilament stimulation (0.16–15g) applied to the centre of the receptive field in MIA-treated and saline-treated rats at day 14 post-injection. There was a significant leftward shift in the stimulus–response curve in MIA-treated rats compared with saline-treated rats (**p<0.01, n=30/37 MIA/saline, respectively). Data are displayed at mean±SEM. Analysis used a two-way ANOVA with Bonferroni's post hoc test. (D) Close intra-arterial administration of the TRPV1 agonist capsaicin (5–10 μM) increased (sensitised) mechanically evoked responses of joint afferents in saline-treated rats. A significant increase in the frequency of firing was observed with 5 μM capsaicin in 40% (8/20) of fibres in saline-treated rats (***p<0.001, n=8). Data are displayed at mean±SEM. Analysis used a two-way ANOVA with Bonferroni's post hoc test. (E) Close intra-arterial administration of capsaicin (5 and 10 μM) significantly reduced mechanical thresholds of 45% (9/20) of joint afferents in saline-treated rats (n=9). Horizontal bars represent median values and analysis used a Mann–Whitney test, *p<0.05.
Is there a functional upregulation of TRPV1 in the joint in the MIA model of OA pain?
In order to focus on the potential role of TRPV1 within the knee joint, we first quantified levels of a known ligand for TRPV1 (12-hydroxy-eicosatetraenoic acid (12-HETE)) in the knee joint and cell bodies of the afferent fibres (DRGs) in MIA-treated rats. 12-HETE was quantified (see online supplementary figure S3) in all the rat knee joints and DRG samples analysed. Levels of 12-HETE were significantly higher in the joint tissue of MIA-treated rats compared with saline-treated rats (p<0.05, figure 2B). These changes were restricted to the joint as levels of 12-HETE in the DRGs were comparable between MIA-treated and saline-treated rats (median: MIA=0.35 pmol/g, range=0.17–1.18 pmol/g; saline=0.23 pmol/g; range=0.07–0.55 pmol/g). TRPV1 mRNA was present in rat synovium expressed at levels that were comparable between saline-treated and MIA-treated rats when adjusted for the housekeeping gene β-actin (see online supplementary figure S4). Given that TRPV1 mRNA expression in synovium is on infiltrating mononuclear cells (eg, macrophages), when levels are adjusted for β-actin, no increase in expression is consistent with increased abundance.
The next experiments investigated whether TRPV1 contributes to the sensitisation of knee joint afferents in the MIA model of OA pain. Mechanically evoked responses of knee joint afferents were significantly increased (figure 2C) and mechanical thresholds were significantly lower in MIA-treated rats (n=30 fibres) compared with saline-treated rats (n=37 fibres) (see online supplementary figure S5). Intra-arterial administration of the TRPV1 agonist capsaicin (5, 10 μM/100 μL) close to the ipsilateral knee joint significantly increased mechanically evoked responses (figure 2D; p<0.001, two-way ANOVA) and reduced mechanical thresholds (figure 2E, Mann–Whitney, p<0.05) of fibres in saline-treated rats, but not MIA-treated rats (data not shown). Intra-arterial administration of the TRPV1 antagonist JNJ-17203212 (0.075 mg and 0.15 mg/100 μL) close to the ipsilateral knee joint inhibited mechanically evoked responses of knee joint afferents (figure 3A; p<0.0001, two-way ANOVA Bonferroni's post hoc tests, n=11) in MIA-treated rats. In addition, the lowered mechanical thresholds of joint afferents in MIA-treated rats were reversed by JNJ-17203212 to the control values seen in saline-treated rats (figure 3C). In saline-treated rats, mechanically evoked responses of afferent fibres (frequency of evoked firing and thresholds) were not altered by JNJ-17203212 (figure 3B, C; n=10). Collectively these data indicate the presence of a tonic activation of joint TRPV1 in the model of OA pain, but not in control rats. Administration of vehicle (2.5% and 5% ethanol, 10% Tween 80 in saline) did not alter mechanically evoked responses of joint afferents in either MIA-treated rats or saline-treated rats (see online supplementary figure S6A,B). Note JNJ-17203212 significantly attenuated capsaicin-evoked knee afferent firing28 (see online supplementary figure S7).

Blockade of peripheral transient receptor potential vanilloid 1 ablates afferent sensitisation in mono-iodoacetate (MIA) model. (A) Close intra-arterial administration of JNJ-17203212 (0.075 mg, 0.15 mg/100 μL) significantly reduced the frequency of mechanically evoked firing of joint afferents in MIA-treated rats (p<0.0001, n=11). (B) By contrast, JNJ-17203212 did not alter mechanically evoked responses of joint afferents in saline-treated rats (n=10). Data are displayed at mean±SEM and analysis used a two-way analysis of variance (ANOVA) with Bonferroni's post hoc test. (C) Mechanical thresholds in MIA-treated rats were significantly lower than saline-treated rats (p<0.001, n=11). Close intra-arterial injection of JNJ-17203212 (0.15 mg/100 μL) reversed the lowered mechanical thresholds of joint afferents in MIA-treated rats (p<0.001, n=11). Mechanical thresholds in MIA-treated rats which received JNJ-17203212 (0.15 mg/100 μL) were not statistically different from control (pre-drug) thresholds in saline-treated rats (n=10). Bars represent median values and analysis used the Kruskal–Wallis test. ns, not significant. *p<0.05, **p<0.01, ***p<0.001.
No functional upregulation of TRPV1 in the spinal cord in the model of OA pain
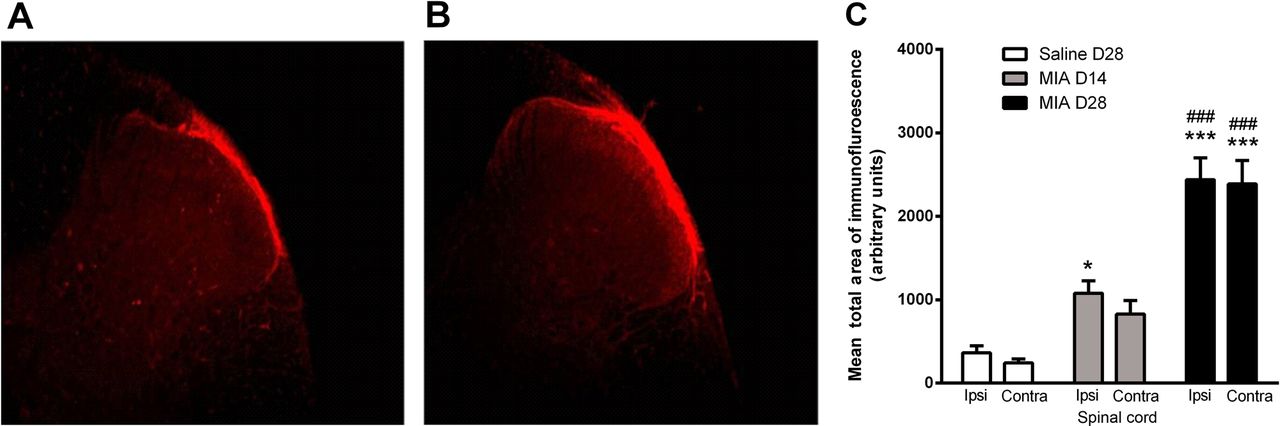
Given the known role of TRPV1 in the spinal processing of noxious inputs and that spinal neuronal responses are facilitated at 28 days in this model of OA,22 the final series of experiments investigated whether spinal TRPV1 is functionally upregulated in this model of OA pain. Consistent with previous reports, TRPV1 immunoreactivity was detected in the superficial laminae (I–II) of the dorsal horn of the spinal cord (figure 4A B). There was a bilateral increase in the mean total area of dorsal horn TRPV1 immunoreactivity in MIA–treated rats, at both days 14 and 28, compared with saline-treated rats (figure 4C). The absence of staining in the negative control confirmed the specificity of detected immunoreactivity.

Transient receptor potential vanilloid 1 (TRPV1) immunoreactivity in the spinal cord. TRPV1 immunofluorescence detected in superficial dorsal horn (10× magnification) in rat lumbar (L3–L5) spinal cord at day 28 post-intra-articular injection of saline (A) or mono-iodoacetate (MIA) (B). Minimum and maximum brightness values were altered (32.01 min and 90.14 max) using Image J so as to highlight the area of TRPV1 positive staining. (C) Quantification of TRPV1 immunofluorescence in superficial dorsal horn of spinal cord taken from rats at 14 or 28 days following intra-articular injection of MIA and at day 28 following intra-articular injection of saline. Data are expressed as mean and SEM (n=5 per group). Analysis used a two-way analysis of variance (ANOVA) with a Bonferroni's post hoc test. *p<0.05 and ***p<0.001 compared with saline treatment and ###p<0.001 between day 14 and day 28 MIA treatment.
Spinal electrophysiological experiments were then undertaken to determine whether the increase in spinal TRPV1 immunoreactivity has a functional consequence on neuronal responses in the MIA model. Spinal administration of JNJ-17203212 (37.5 and 75 µg) produced a similar dose-related inhibition of noxious (15, 26 and 60 g) mechanically evoked responses of dorsal horn WDR neurones in saline-treated rats (figure 5A) and MIA-treated rats (at 14 and 28 days) (figures 5B,C). Spinal administration of vehicle did not significantly alter mechanically evoked responses of WDR neurones in day 28 MIA-treated rats (figure 5D). These data suggest that despite the increased expression of TRPV1 in the spinal cord, there is no increase in the contribution of spinal TRPV1 to noxious-evoked responses of spinal neurones in the model of OA pain. In keeping with this finding, levels of the endogenous TRPV1 ligand 12-HETE were comparable in the ipsilateral spinal cords of MIA-treated and saline-treated rats (median: MIA=17.4 pmol/g; range=5.65–33.58 pmol/g; saline=15.78 pmol/g, range=8.56–23.13 pmol/g, n=8 per group).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Effect of spinal JNJ-17203212 on evoked responses of wide dynamic range (WDR) neurones. JNJ-17203212 significantly and dose-relatedly inhibited noxious mechanically evoked responses of dorsal horn WDR neurones (15, 26 and 60g) in saline-treated rats (n=7 rats) at 28 days post-injection (A) and in mono-iodoacetate (MIA)-treated rats at 14 (n=9 rats) and 28 days (n=8 rats) post-injection (B and C). No significant effect of the drug was noted on non-noxious mechanically evoked responses (8 and 10g) in any of the treatment groups. (D) Spinal administration of vehicle did not significantly affect mechanically evoked responses in day 28 MIA-treated rats (n=7 rats). Data are displayed at mean±SEM. Analysis used two-way analysis of variance (ANOVA) with Bonferroni's post hoc test; *p<0.05, **p<0.01 and ***p<0.001.
Discussion
Here we report new clinical evidence for a functional upregulation of joint TRPV1 associated with OA, and preclinical evidence that this channel contributes to altered pain responses in an experimental model of OA. Our data from patients with OA knee pain undergoing TKR surgery demonstrate an increase in TRPV1 protein both in OA and in RA synovia compared with non-arthritic PM controls. This increased expression of TRPV1 protein was associated with macrophage infiltration, indicating that non-neuronal cells may contribute a large proportion of synovial TRPV1. Preclinical studies revealed new evidence that joint levels of an endogenous ligand for TRPV1, 12-HETE, were elevated in this model of OA pain and that local pharmacological blockade of TRPV1 attenuated OA-induced joint afferent sensitisation and pain responses. The outcomes of these electrophysiological and behavioural studies support our hypothesis that targeting TRPV1 in the diseased joint may provide an alternative strategy for treating OA pain in the absence of changes in core body temperature.
Clinical studies measuring pain pressure thresholds report sensitisation of the OA joint.5 ,6 ,29 Clinically relevant animal models of OA are essential for the identification of the mechanisms which lead to this sensitisation seen clinically. As well as mimicking the common features of human OA joint pathology and associated pain behaviour,21 ,22 the MIA model of OA also exhibits sensitisation of joint afferents.24 ,30 ,31 Here we report new evidence that peripheral TRPV1 has a functional role in the maintenance of OA-induced joint afferent sensitisation, which is consistent with the role of TRPV1 in afferent mechanical sensitisation during visceral32 and cutaneous inflammation.33 Local blockade of TRPV1 reversed the sensitisation of knee joint afferents, restored evoked responses of these afferents to control values in the model of OA pain and attenuated pain behaviour (weight-bearing asymmetry) to an extent comparable with the effect of systemic injection of the TRPV1 antagonist. These novel findings suggest that the knee joint is an important site of action of TRPV1 antagonists and may contribute to the inhibitory effects of systemic TRPV1 antagonists on pain responses in models of OA.8–11
Multiple mechanism(s), including increased expression and/or sensitisation of TRPV1 and increased levels of endogenous ligands, may lead to a heightened contribution of peripheral TRPV1 to somatosensory processing in OA. The increase in abundance of TRPV1 protein in the synovium from OA patients undergoing TKR was predominantly colocalised with CD68, a marker for macrophages, which precluded the characterisation of possible changes in TRPV-1 abundance in sensory nerves within the synovium itself. TRPV1 expression by mononuclear cells and fibroblasts has previously been reported34 and this finding is consistent with increased macrophage infiltration in OA synovium.26 Collectively our data suggest that the increased TRPV1 abundance in arthritic synovium results from inflammatory cell hyperplasia rather than increased expression by resident synovial cells. Post-transcriptional modifications of TRPV1 may also alter protein turnover35 and contribute to the altered expression of TRPV1 protein in arthritic synovium. The expression of TRPV1 by macrophages in OA is likely to produce proinflammatory effects,34 similar to those reported for OA synovial fibroblasts where TRPV1 activation promotes the release of interleukin 6,36 which can sensitise joint afferent fibres.37 Clinical data suggest that increased expression of TRPV1 may be associated with synovitis, which is a feature of human OA and the MIA model of OA. The precise contribution and mechanisms by which synovitis may mediate OA pain remain uncertain, and the balance between inflammatory and non-inflammatory mechanisms may differ between patients and over time. As such, peripheral analgesic actions of TRPV1 antagonists may be most pronounced in patients whose pain is associated with synovitis. Selective recruitment of patients without an inflammatory component to their pain into clinical trials of TRPV1 antagonists may conceal potential benefits in other patient groups.
TRPV1 protein expression is upregulated in the cell bodies of afferent fibres innervating the joint in a model of OA pain13 and can be activated by a wide range of endogenous bioactive lipids.7 ,38 12-HETE is an endogenous TRPV1 ligand known to increase the firing of afferent fibres via TRPV1 activation.39 Here we report that endogenous levels of 12-HETE are elevated in the knee joint, but not the DRGs or spinal cord, in the MIA model of OA pain, providing a plausible mechanism by which TRPV1 function is facilitated at this level. Further, our RT-PCR data indicate increased TRPV1 mRNA expression in the synovium following MIA. These findings and the demonstration that intra-articular injection of a TRPV1 antagonist blocks established OA pain behaviour (weight-bearing asymmetry) in the MIA model of OA strongly support peripheral TRPV1 making an important functional contribution to the maintenance of OA pain responses. It is well established that systemic administration of TRPV1 antagonists attenuates pain responses in the MIA model8–11 but at the expense of on-target-mediated hyperthermia,10 ,40 which may influence therapeutic potential. Here we demonstrate that intra-articular administration of an analgesic dose of the TRPV1 antagonist does not alter core body temperature, suggesting that the localised delivery of this class of drugs may afford therapeutic benefit in the absence of hyperthermia.
As well as playing a role as an integrator of noxious stimuli in the periphery, TRPV1 is present on the central endings of the sensory afferents in the spinal cord and contributes to spinal nociceptive processing.41 ,42 Analytical and pharmacological evidence presented herein indicates that the functional role of spinal TRPV1 is unaltered in the model of OA pain, despite increased TRPV1 immunoreactivity in the superficial laminae of the dorsal horn in the model of OA pain. The bilateral increase in TRPV1 expression suggests contralateral activation at the spinal level following MIA, consistent with previous findings that unilateral arthritis can induce bilateral changes in spinal neuronal phenotype.43
In summary, the increased abundance of TRPV1 in our clinical arthritic synovium samples along with the polymorphisms in TRPV1 associated with human OA pain12 emphasise the relevance of TRPV1 to human OA pain. Our novel experimental evidence for increased functional role of TRPV1 at the level of the joint in a model of OA pain and the demonstration that blockade of joint TRPV1 ablates sensory afferent sensitisation and pain behaviour support future targeted site-specific investigations of the therapeutic potential of TRPV1 for OA pain associated with synovitis.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
SK and RJC are joint first authors
Contributors SK, VC, RJC, SW, JJB, DRS, DAW, DFM, AW, GH and JT were involved in the conception and design of the studies. All authors were involved in the analysis and interpretation of data, drafting the article, revising it critically for important intellectual content and final approval of the version to be published.
Funding This work was funded by Arthritis Research UK, grant number 18769.
Competing interests None.
Ethics approval North Nottinghamshire local research ethics committee.
Provenance and peer review Not commissioned; externally peer reviewed.