Article Text

Abstract
Objective To assess the 1-year efficacy and safety of a regimen of tocilizumab plus methotrexate or placebo, which was augmented by a treat-to-target strategy from week 24.
Methods ACT-RAY was a double-blind, 3-year trial. Adults with active rheumatoid arthritis despite methotrexate were randomised to add tocilizumab to ongoing methotrexate (add-on strategy) or to switch to tocilizumab plus placebo (switch strategy). Tocilizumab 8 mg/kg was administered every 4 weeks. Conventional open-label disease-modifying antirheumatic drugs (DMARDs) other than methotrexate were added at week 24 or later in patients with DAS28>3.2.
Results 556 patients were randomised; 85% completed 52 weeks. The proportion of patients receiving open-label DMARDs was comparable in the add-on (29%) and switch (33%) arms. Overall, week 24 results were maintained or further improved at week 52 in both arms. Some endpoints favoured the add-on strategy. Mean changes in Genant-modified Sharp scores were small; more add-on (92.8%) than switch patients (86.1%) had no radiographic progression. At week 52, comparable numbers of patients had antidrug antibodies (ADAs; 1.5% and 2.2% of add-on and switch patients, respectively) and neutralising ADAs (0.7% and 1.8%). Rates of serious adverse events and serious infections per 100 patient-year (PY) were 11.3 and 4.5 in add-on and 16.8 and 5.5 in switch patients. In patients with normal baseline values, alanine aminotransferase elevations >3× upper limit of normal were observed in 11% of add-on and 3% of switch patients.
Conclusions Despite a trend favouring the add-on strategy, these data suggest that both tocilizumab add-on and switch strategies led to meaningful clinical and radiographic responses.
- Rheumatoid Arthritis
- DMARDs (biologic)
- Methotrexate
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
The goal of rheumatoid arthritis (RA) therapy is to reduce or prevent functional impairment and structural damage that can occur over a patient's lifetime. Long-term control is often best achieved through the adaptation of treatment based on disease activity (treat-to-target).1 Typical treatment modifications include the addition of conventional disease-modifying antirheumatic drugs (DMARDs) to pre-existing therapy, the addition of a biologic to a conventional DMARD or a switch from a conventional DMARD to a biologic therapy.
When disease control is inadequate with the first traditional DMARD, typically methotrexate, patients will frequently receive biologic therapy in addition to or instead of methotrexate.2 In clinical practice, approximately one-third of patients with RA are being treated with biologic monotherapy,3–5 often because of tolerability issues with methotrexate.6–8 One such biologic therapy is tocilizumab, a humanised antihuman interleukin-6 (IL-6) receptor monoclonal antibody.9
Tocilizumab is efficacious and generally well tolerated in a wide range of patients with RA when given as either monotherapy10 or in combination with methotrexate11 and other DMARDs.12 Long-term studies have demonstrated that tocilizumab can reduce the signs and symptoms of RA for several years in combination with a conventional DMARD and as monotherapy.10 ,13–15 Further, tocilizumab, in combination with methotrexate, has been shown to inhibit radiographic progression for up to 3 years in patients with an inadequate response to methotrexate.14
ACT-RAY is a 3-year, phase 3b, randomised, double-blind clinical trial. The first 24 weeks of ACT-RAY assessed the efficacy and safety of adding tocilizumab to ongoing methotrexate (add-on strategy) versus switching to tocilizumab monotherapy (switch strategy) in patients with moderate to severe active RA experiencing an inadequate response to methotrexate.16 The primary efficacy analysis of the study at week 24 did not succeed at demonstrating superiority of the add-on over the switch strategy, suggesting that switching to tocilizumab monotherapy might be a valuable treatment strategy for patients for whom methotrexate is contraindicated or poorly tolerated.16
From week 24 to year 3, ACT-RAY employed a treat-to-target strategy. During weeks 24–52, patients continued on tocilizumab therapy with blinded methotrexate or placebo (PBO), but open-label conventional DMARDs were added based on disease activity, with the ultimate objective to induce clinical remission during the course of the study. This article reports on the main objectives of the 52-week analyses, which were to evaluate the sustainability of the response observed at week 24 and to further evaluate the treatment strategies in terms of clinical activity, structural damage, immunogenicity and changes in concomitant therapies (all secondary study objectives). Full assessment of treatment adaptation strategies (step-up and step-down) will occur after year 2.
Patients and methods
This report covers the planned analysis for the first 52 weeks of a 3-year, double-blind, PBO-controlled, parallel-group clinical trial (NCT00810199, EudraCT No. 2008-001847-20). The study was approved by the appropriate institutional review boards/ethics committees and was conducted in accordance with International Conference on Harmonisation Good Clinical Practice and local regulations. All patients provided written informed consent for the 3 years of the ACT-RAY study.
Patients and methods have been previously described.16 Briefly, patients had confirmed RA according to the 1987 American College of Rheumatology (ACR) classification criteria with Disease Activity Score based on 28 joints-erythrocyte sedimentation rate (DAS28-ESR) >4.4 at baseline, despite a stable methotrexate dose of at least 15 mg/week for >6 weeks and radiographic evidence of RA-related joint erosions. Major exclusion criteria included previous use of biologics and any conventional DMARD treatment other than methotrexate during the month preceding baseline visit. At randomisation, patients either added open-label tocilizumab 8 mg/kg intravenously every 4 weeks to their existing methotrexate (add-on strategy) or were switched to tocilizumab alone (switch strategy; with PBO). Until week 24, these treatments were maintained except for safety reasons.
Treat-to-target and concomitant RA treatments
Following the primary endpoint analysis at week 24, treatment was intensified based on individual disease activity while blinding was maintained in both study groups. At week 24, if a patient had a DAS28 >3.2, an open-label conventional DMARD (sulfasalazine, leflunomide, chloroquine, hydroxychloroquine, parenteral gold or azathioprine; choice and dose at investigator's discretion) was added. At week 36, if a patient still had a DAS28 score >3.2 with the added DMARD, the patient was moved to the maintenance-regimen arm with the option of adding an additional conventional DMARD at the investigator's discretion. Patients in the maintenance arm did not undergo systematic step-up or step-down therapy but were treated as per the investigator's discretion. Tocilizumab and/or DMARD treatment was reduced or temporarily interrupted in patients with alanine aminotransferase (ALT) or aspartate aminotransferase (AST) values >1 to 3× the upper limit of normal (ULN) and discontinued for persistent increases >3× ULN (see online supplementary table S2 for risk mitigation strategy).
Oral corticosteroids (≤10 mg/day prednisone or equivalent) and non-steroidal anti-inflammatory drug (NSAIDs) were permitted, provided that doses remained stable for at least 25 of 28 days prior to the start of study treatment. Alterations in NSAID dose were not recommended during the study, particularly during the first 24 weeks. The corticosteroid dose could not be changed during the first 24 weeks of the study. From week 24, if DAS28 ≤3.2, corticosteroid treatment could be reduced by 2.5 mg/day prednisone-equivalents at week 24 or any visit thereafter; however, the dose may not have been reduced to <5 mg/day during the first year or be increased back to a level above the patient's baseline dose.
Collected patient data and assessments
Efficacy, radiographic and safety assessments
Clinical and laboratory data were collected every 4 weeks.16 Radiographs of hands/wrists and feet were obtained at baseline, week 24 and week 52 and were assessed by applying the Genant-modified Sharp score (GSS) from two independent readers (Perceptive Informatics Medical Imaging Services, Berlin, Germany) who were blinded to treatment assignment, chronological order of radiographs and patient's clinical status. The smallest detectable change (SDC)17 for GSS was computed based on the observed SD of difference between the X-ray readers; three readers in total participated in the campaign.
Immunogenicity
Following a regulatory request regarding the tocilizumab development programme, immunogenicity testing was introduced to ACT-RAY while the study was ongoing. Patients were tested for antidrug and neutralising antibodies using available samples from baseline, week 24 and week 52 (or withdrawal visit not later than week 52). Samples were analysed using a screening assay first. If positive for antitocilizumab antibodies, a neutralising assay was performed to test for the ability of those antibodies to inhibit the activity of tocilizumab. The cut-off for assay positivity was determined using baseline sera from ACT-RAY patients.
Statistical analysis
The target sample size was 235 patients per arm, resulting from power considerations around the primary analysis of the week 24 DAS28 remission rate.16 For this and similar endpoints, logistic regression models were employed including region and baseline DAS28 or the respective baseline values (eg, CDAI or radiographic scores), with supportive Cochran–Mantel–Haenszel tests stratified for the same parameters. Analysis of covariance models with similar adjustments was used to compare continuous outcome measures. For the analyses presented here, statistical significance was declared for p values <0.05 and no correction for multiplicity was employed.
Efficacy was analysed in the intention-to-treat (ITT) population using non-responder imputation for categorical variables (eg, DAS28 remission, ACR response and radiographic progression). The primary endpoint has been previously reported.16 Efficacy endpoints were analysed in accordance with the European League Against Rheumatism (EULAR)/ACR collaborative recommendations for reporting RA disease activity in clinical trials18 and included the mean change in DAS28-ESR, ACR20/50/70/90 response and ACR core components. Radiographic endpoints included changes from baseline in total GSS, erosion and joint space narrowing scores, and the proportion of patients with no radiographic progression (progression defined as change in GSS >1.5 for this specific reading campaign). The means were adjusted for region, baseline DAS28 and GSS, and the 52-week analysis used linear extrapolation if the week 24 assessment was evaluable. Safety endpoints included the incidence of adverse events (AEs), serious adverse events (SAEs), serious infections (SIs) and specific laboratory abnormalities, which were analysed in the safety population (all treated patients with at least one postdose assessment of safety, analysed according to the received treatment).
Results
Patient flow and baseline characteristics
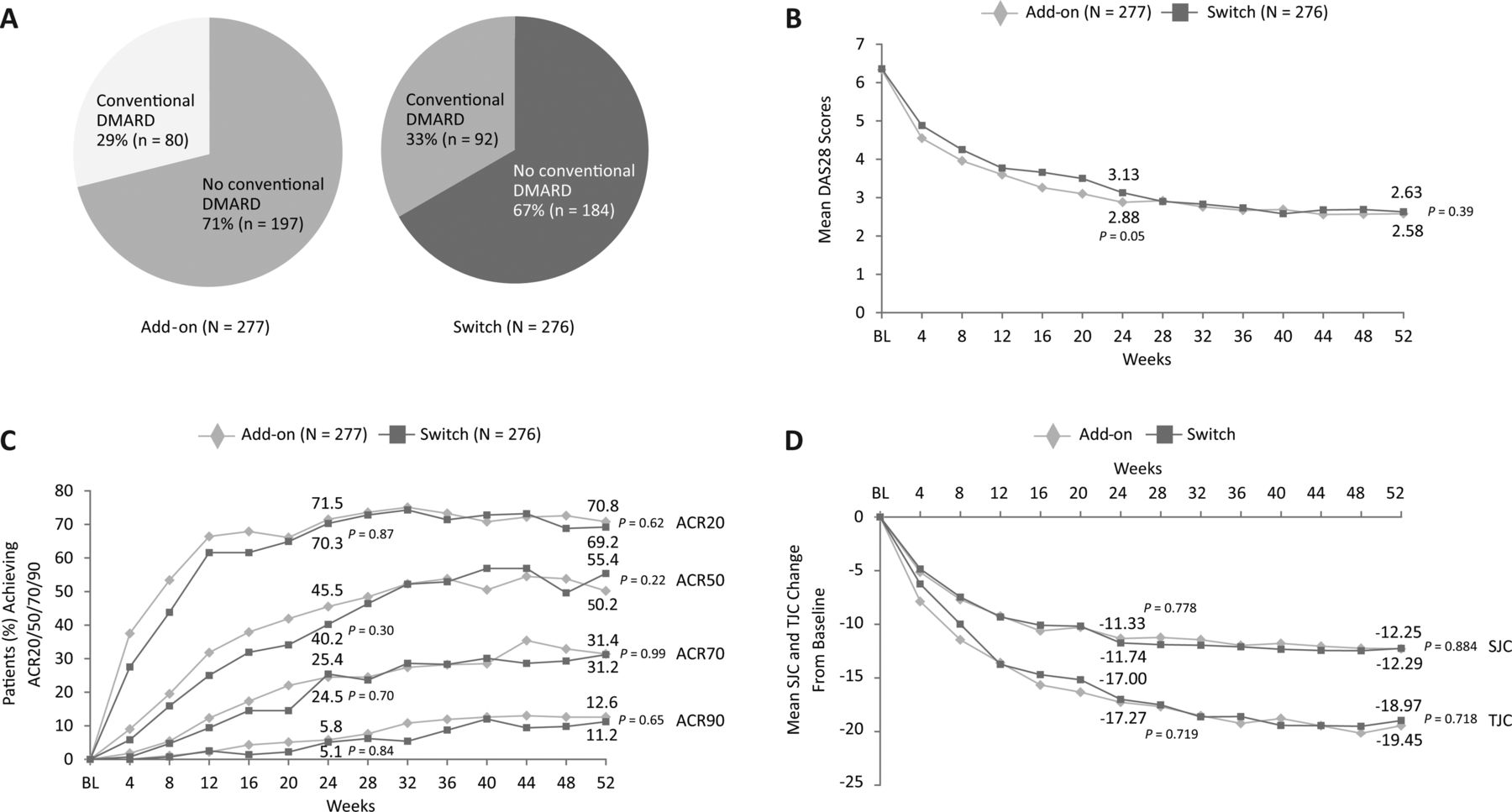
Figure 1 summarises the patient disposition through 52 weeks. In total, 556 patients were recruited, exceeding the target of 470 patients. Of the 556 patients, 509 (92%) completed the first 24 weeks of the trial and 472 (85%) completed 52 weeks. More patients discontinued the study due to a lack of efficacy in the switch arm (12 patients) versus the add-on arm (5 patients). Both patient groups were well matched for baseline characteristics, except for a difference in GSS (which did not affect outcomes; table 1).
Baseline characteristics

Patient disposition and study flow chart. ITT, intention-to-treat; MTX, methotrexate; PBO, placebo; TCZ, tocilizumab.
Clinical efficacy
Sustainability of therapeutic response
Table 2 summarises the efficacy results by treatment group at weeks 24 and 52, and key measures are depicted over time in figure 2B–D. The improvements in signs and symptoms of RA achieved by week 24 were maintained or further improved up to week 52 in both the add-on and the switch groups. Overall, no clinically relevant difference between the two groups was observed. However, some results favoured the add-on strategy, as the percentage of patients with DAS28 remission (DAS28 <2.6, 45.5% vs 36.6%), the change in patient's global assessment of pain, the change in erosion score and the percentage of patients with no progression in GSS showed statistically significant differences in favour of the add-on strategy. Other study endpoints, including mean change in DAS28, did not result in statistically significant differences between the study arms.
Efficacy results at weeks 24 and 52

Efficacy results. (A) Percentage of patients receiving additional conventional disease-modifying antirheumatic drugs (DMARDs) between weeks 24 and 52. Results over time for (B) mean DAS28, (C) percentage of patients achieving ACR20, ACR50, ACR70 and ACR90, and (D) mean change from baseline in SJC and TJC. ACR, American College of Rheumatology; DAS28, Disease Activity Score based on 28 joints; DMARD, disease-modifying antirheumatic drug; TJC, tender joint count; SJC, swollen joint count. p values are for week 24 and week 52 intergroup data, respectively.
Measures specifically tracking the sustainability of response were also examined. Sustained ACR20 response was defined as at least six consecutive visits during a 1-year period in which a patient had an ACR20 response. At week 52, 138 patients (49.8%) and 131 patients (47.5%) from the add-on group and switch groups, respectively, achieved a sustained ACR20 response (p=0.5948). The percentage of patients in the add-on group who achieved sustained low-disease activity state (DAS28≤3.2 at three or more consecutive visits) was similar to that in the switch group (46.9% vs 41.3%, respectively; p=0.171). Further, the percentage of patients in the add-on group who achieved sustained DAS28 remission (DAS28<2.6 at three or more consecutive visits) was similar to that in the switch group (28.5% vs 25.4%, respectively; p=0.351).
Structural outcomes
At 52 weeks, the majority of patients exhibited no or minimal progression of structural damage (figure 3C). There was no statistically significant intergroup difference in mean change from baseline at week 24 or week 52 for the mean total GSS or the joint space narrowing score (table 2 and figure 2A). The adjusted mean change in erosion score was −0.09 for add-on patients and 0.25 for switch patients (p=0.001), which followed a statistically significant difference at week 24 (table 2 and figure 2B). The SDC from baseline in GSS was 1.5, indicating a high agreement of the readings, therefore allowing for detection of changes from baseline <−1.5 and >1.5. When radiographic progression was defined as any change in GSS >SDC, 92.8% of add-on patients and 86.1% of switch patients did not exhibit radiographic progression at week 52 (p=0.016) (figure 3B). When the cut-off for radiographic progression was defined as change in GSS ≥ 0, 68.8% of add-on patients and 63.6% of switch patients did not exhibit radiographic progression at week 52.

{kind=link}
{kind=link}
{kind=link}
Radiographic results. (A) Mean change from baseline in total Genant-modified Sharp score (GSS), (B) patients without radiographic progression and (C) cumulative distribution plot of change from baseline to week 52 in total GSS. Area between grey lines is within SDC. GSS ≤1.5; patients without an assessment classified as progressors, p values from Cochran–Mantel–Haenszel χ2 tests with strata defined by baseline score quartile and baseline DAS28 ≤ or >5.5; error bars=SE of the mean; baseline annualised progression rate was GSS divided by RA duration. DAS28, Disease Activity Score based on 28 joints; GSS, Genant-modified Sharp score; RA, rheumatoid arthritis. SDC, smallest detectable change.
DMARD and corticosteroid use
A comparable proportion of patients in both treatment arms received additional conventional DMARDs at week 24 or later (figure 2A; see online supplementary table S1). Some patients with DAS28 just above 3.2 at week 24 did not add DMARDs. Had the protocol been strictly followed, approximately 4% more of the add-on patients and 9% more of the switch patients would have added conventional DMARDs at week 24. By week 52, a total of 38 (14%) of add-on patients and 49 (18%) of switch patients had added a further DMARD. Further, by week 52, the mean corticosteroid dose had changed from 6.8 mg/day at baseline to 6.4 mg/day in the add-on arm and from 6.7 mg/day at baseline to 6.3 mg/day in the switch arm.
Safety
Overall safety
An overview of the safety results is presented in table 3. Overall, rates per 100 patient-years (PYs) of AEs, SAEs and SIs were similar between groups. SAEs occurring in more than one patient were congestive heart failure (0.7% (two patients) and 0.4% (one patient) in the add-on and switch groups, respectively), tendon rupture (0% and 0.7% (two patients), respectively), pneumonia (0% and 0.7% (two patients), respectively), septic shock (0.7% (two patients) and 0%, respectively), increased transaminases (0.4% (one patient) in both groups), anaemia (0.4% (one patient) in both groups) and anxiety (0.4% (one patient) in both groups). Additionally, there were four deaths in the first 52 weeks of the study, two in each study arm. The causes of death in these four patients were (1) sepsis (add-on group); (2) septic shock preceded by scrotal abscess, skin necrosis, acute renal failure and congestive heart failure (add-on group); (3) myocardial infarction (switch group) and (4) sepsis with meningitis (switch group).
Overview of adverse events, deaths, liver enzyme elevations and antitocilizumab antibodies until week 52
Through week 52, SIs had occurred at the rate of 4.5 and 5.5 per 100 PYs for the add-on and switch groups, respectively. From baseline to week 24, the SI rate was 6.27 and 5.61 for the add-on and switch groups, respectively; and 2.88 and 5.33 from week 24 to week 52.
Immunogenicity
Valid immunogenicity data (baseline and postbaseline) were available for 86.3% of add-on patients (239) and 80.8% (223) of switch patients. The rate of overall and neutralising antidrug antibodies (seroconversion in neutralising assay) was similar in both treatment groups (table 3).
Discussion
At week 52 of the ACT-RAY study, the analyses confirmed the sustainability of the suppression of disease activity observed at week 24 with tocilizumab when used either as monotherapy after discontinuing methotrexate or in combination with methotrexate (with or without the addition of open-label DMARDs).
The first 24 weeks of the ACT-RAY study compared the tocilizumab add-on and switch-to-monotherapy strategies in patients with inadequate or incomplete response to methotrexate. Week 24 marked the beginning of the treat-to-target concept, with treatment changes determined by disease activity. Because of the introduction of conventional DMARDs, the week 52 results do not represent a pure assessment of tocilizumab monotherapy versus combination therapy with methotrexate but include the growing effects of a treat-to-target strategy consistently applied to both study arms.
The changes in non-tocilizumab therapies from week 24 complicate the interpretation of the switch versus the add-on concept. The disease activity-dependent addition of open-label conventional DMARDs (of the investigator's choice) from week 24 was mandated by the study protocol. In the absence of an appropriate control and because the addition of the DMARD was possible at different times, the effectiveness of this intervention cannot be properly assessed and isolated from other effects, such as late response to the study treatments. Reducing the corticosteroid dose was also possible but only within predefined limits to minimise any between-group differences that corticosteroid dose reductions could have on structural outcomes. Both the addition of conventional DMARDs (other than methotrexate) and the dose reduction of corticosteroids occurred in similar proportions of patients in the add-on and switch arms.
Improvements in signs and symptoms of RA observed at week 24 were maintained or further improved up to week 52 in both treatment groups. Results pointed towards a slightly more favourable response for the add-on arm as numerical trends were observed for most clinical and radiographic endpoints. In exploratory analyses, statistical significance in favour of the add-on arm was identified for 4 of 18 endpoints, including clinical and radiographic endpoints.
The addition of DMARDs according to disease activity in both arms was expected to lead to a ‘dilution’ of potential distinctions between the two arms, resulting in smaller differences at week 52 than at week 24, as seen with other treat-to-target approaches.19 However, for the progression of structural damage, a difference was hardly visible at week 24 but emerged more clearly by week 52, possibly suggesting methotrexate as a causative factor. On the other hand, the cumulative distribution plot (figure 3C) demonstrates that, overall, only a few patients benefited from continuously receiving methotrexate.
Antidrug antibodies and neutralising antibodies were detected in similar proportions of patients in the two groups. The interpretation of this analysis is limited by the fact that the immunogenicity testing was introduced after the trial had started, and not all patients could be included. As antidrug antibody positivity rates are very low compared with other biologics,20–22 these data suggest that the immunogenic potential of tocilizumab could be low. However, these results do not exclude the risk of serious hypersensitivity reactions.23
Other safety outcomes were consistent between the two study groups, except that the add-on strategy resulted in a numerically higher proportion of patients who had transaminase level increases compared with the switch strategy. This trend was also observed at week 24 and in other tocilizumab clinical trials, including ACT-STAR and ACT-SURE.16 ,24 ,25 These observations suggest that methotrexate and tocilizumab may have an additive effect on liver transaminases, the clinical relevance of which remains unclear. Overall, the incidence of events such as SAEs, SIs and discontinuations was similar between the strategies and consistent with previous tocilizumab studies.
Both the add-on and the switch strategies (with or without additional DMARDs) led to meaningful clinical and radiographic responses that could be maintained and, in some cases, improved upon in the first year of treatment. While there was no major overall difference between the add-on and switch treatment strategies, ACT-RAY provides useful information for clinical decision making on a per-patient basis. For example, the addition of tocilizumab to methotrexate may be the preferred approach in patients who can tolerate methotrexate as per EULAR treatment recommendations.26 However, for patients who cannot be treated with methotrexate, a switch to tocilizumab monotherapy is an option that may provide a robust level of disease control. Examination of other sets of patients with RA (including registries) will be necessary to confirm and further corroborate the results reported herein.
Acknowledgments
The authors acknowledge and thank Paul-Peter Tak for his critical contributions to the design of the study. Support for third-party writing assistance by Health Interactions, San Francisco, California, USA, for this manuscript was provided by F. Hoffmann-La Roche Ltd.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online supplement
Footnotes
Handling editor Tore K Kvien
-
Contributors MD, KK, PGC, EMM, GS, CB and TWJH designed the study and analysed and interpreted the data. MD, PGC, EMM, GS, RG, MSH, HA, RX, OT and TWJH, along with other investigators, were involved in generating the data at their clinical research sites. All authors were involved in writing the manuscript and approved it.
-
Funding The ACT-RAY study was funded by F. Hoffmann-La Roche Ltd., Basel, Switzerland (Roche). Roche was involved in developing the study design, in the analysis and interpretation of the data, in the writing of the report and in the decision to submit the paper for publication, through Roche employees, contractors and funding of third-party support, such as contract research organisations. All these activities happened in close collaboration with the independent members of the Scientific Steering Committee.
-
Competing interests MD has been a consultant for Abbott, Pfizer, Roche and UCB. KK is an employee of Roche Products Pty Ltd. PGC has participated in speakers’ bureaus for BMS, Janssen, Pfizer, Roche. MSH has been a consultant for Abbott and Roche. RX has been a consultant for AstraZeneca, Janssen, Merck, Pfizer and Roche. OT was granted research support from the ACT-RAY clinical trial and has been a consultant for Abbott, Amgen, BMS, Centocor, Pfizer, Roche and Takeda. CB is a contractor of F. Hoffmann-La Roche Ltd. TWJH has been a consultant for Abbott, Axis Shield Diagnostics, Biotest AG, BMS, Crescendo Bioscience, Novartis, Pfizer, Roche, Schering-Plough, UCB and Wyeth.
-
Patient consent Obtained.
-
Ethics approval Appropriate institutional review boards/ethics committees.
-
Provenance and peer review Not commissioned; externally peer reviewed.
Linked Articles
- Clinical and epidemiological research