Article Text

Abstract
Objectives To assess whether combination therapy with infliximab (IFX) plus nonsteroidal anti-inflammatory drugs (NSAIDs) is superior to NSAID monotherapy for reaching Assessment of SpondyloArthritis international Society (ASAS) partial remission in patients with early, active axial spondyloarthritis (SpA) who were naïve to NSAIDs or received a submaximal dose of NSAIDs.
Methods Patients were randomised (2 : 1 ratio) to receive naproxen (NPX) 1000 mg daily plus either IFX 5 mg/kg or placebo (PBO) at weeks 0, 2, 6, 12, 18 and 24. The primary efficacy measure was the percentage of patients who met ASAS partial remission criteria at week 28. Several other measures of disease activity, clinical symptoms and patient-rated outcomes were evaluated. Treatment group differences were analysed with Fisher exact tests or analysis of covariance.
Results A greater percentage of patients achieved ASAS partial remission in the IFX+NPX group (61.9%; 65/105) than in the PBO+NPX group (35.3%; 18/51) at week 28 (p=0.002) and at all other visits (p<0.05, all comparisons). Results of most other disease activity and patient-reported endpoints (including Ankylosing Spondylitis Disease Activity Score, Bath Ankylosing Spondylitis Disease Activity Index, Bath Ankylosing Spondylitis Functional Index, multiple quality of life measures and pain measures) showed greater improvement in the IFX+NPX group than the PBO+NPX group, with several measures demonstrating early and consistent improvement over 28 weeks of treatment.
Conclusions Patients with early, active axial SpA who received IFX+NPX combination treatment were twice as likely to achieve clinical remission as patients who received NPX alone. NPX alone led to clinical remission in a third of patients.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/
Statistics from Altmetric.com
Introduction
The term axial spondyloarthritis (SpA) is the umbrella term for patients with ankylosing spondylitis (AS) according to the modified New York criteria1 and for patients who do not yet show signs of structural damage in the sacroiliac (SI) joint that are visible as radiographic sacroiliitis and, therefore, categorised as nonradiographic axial SpA. Recently, new classification criteria for axial SpA have been developed, which cover both subgroups.2 There are currently only two treatments with proven efficacy available for these patients with axial SpA: nonsteroidal anti-inflammatory drugs (NSAIDs) and tumour necrosis factor (TNF)-α–targeted therapy.3 Until now, TNF antagonists have only been investigated and are only recommended for patients with axial SpA who fail previous NSAID therapy.3 ,4 Although data are limited, studies have demonstrated up to 50% remission rate with TNF-α antagonist therapy in NSAID-refractory patients with axial SpA who are treated in the first 3–5 years of their disease.5–7 This raises the question of whether even earlier treatment of axial SpA in patients who are not refractory to NSAID therapy would result in even higher response rates and potentially even in biologic drug-free remission, as has been recently investigated in great detail in patients with rheumatoid arthritis.8 ,9
The Infliximab (IFX) as First Line Therapy in Patients with Early Active Axial Spondyloarthritis Trial (INFAST) evaluated whether combination therapy with the TNF antagonist IFX and naproxen (NPX) was superior to treatment with NPX alone in patients who had active moderate-to-severe axial SpA and who were naïve to NSAIDs or had only been treated with a submaximal dose of NSAIDs. All patients had to fulfil the Assessment of SpondyloArthritis international Society (ASAS) classification criteria for axial SpA, thus including patients both with AS and with nonradiographic axial SpA; had to have a disease duration ≤3 years; and had to have evidence of inflammatory SI lesions on MRI at baseline. Thus, this study is the first investigation of the potential benefits of early TNF-antagonist treatment in active axial SpA patients who are not yet refractory to NSAID therapy.
Methods
Design and patients
INFAST was a Phase 3b, randomised, parallel-group, multisite, double-blind, placebo (PBO)-controlled study of IFX in adults with moderate-to-severe, active axial SpA who were not refractory to NSAIDs (Protocol P05336, NCT00844805). Patients were recruited consecutively by rheumatologists in hospitals or private practice settings. Patients were enrolled in 47 centres in nine countries (Austria, Belgium, Denmark, France, Germany, Hungary, Russia, South Korea and Ukraine). The study protocol was reviewed by appropriate institutional review boards for each study site. All patients gave written informed consent to participate. Data were collected between 22 October 2009, and 20 September 2011.
Patients were 18–48 years of age with a diagnosis of active axial SpA according to the local investigator and disease duration of ≤3 years. All patients had to fulfil the imaging portion of the ASAS criteria for axial SpA,2 with active inflammation of the SI joints (defined as bone oedema within or adjacent to the SI joints) as shown by short tau inversion recovery MRI. For inclusion into the study, the MRI scans were read locally. All patients had active disease at screening and baseline, defined as a total back pain evaluation of ≥40 mm (visual analogue scale (VAS) of 0–100 mm) and a Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score of ≥4 cm (0–10 cm VAS). Patients were either NSAID-naïve at baseline or had been treated with not more than two-thirds of the maximal recommended dose10 during the 2 weeks prior to screening and had undergone a washout period of ≥3 days before baseline, during which they had an increase in total back pain of ≥30%.
Study treatment
During Part 1 of INFAST patients were randomised in double-blind fashion at a 2 : 1 ratio to receive either intravenous (IV) IFX 5 mg/kg or IV PBO at weeks 0, 2, 6, 12, 18, and 24 (see online supplementary figure S1). Both groups also received oral NPX 1000 mg daily. A computer-generated randomisation list was created by the sponsor and held by the central randomisation centre, which was contacted by the site to assign treatment to each patient as he or she enrolled. Patients who met ASAS partial remission criteria at week 28 were eligible to participate in Part 2 of INFAST, which compared maintenance of partial remission with two follow-up regimens (NPX alone or no treatment); this portion of the study is reported separately.
Outcome measures
The primary efficacy measure was the percentage of patients in each treatment group who met ASAS partial remission criteria at week 28. A number of secondary measures of disease activity, clinical signs and symptoms, inflammatory markers, and patient-reported outcomes were also assessed.
Adherence to NPX treatment was measured as the percentage of days in the study that the daily dose was taken as reported on patient diary cards. Adherence to IFX and placebo was measured as the number of doses infused of the number of scheduled doses.
Adverse events (AEs) and several other safety measures were also collected.
Statistical analyses
The targeted sample size was 150 patients (100 receiving IFX+NPX and 50 receiving PBO+NPX) for 90% power to detect a 30% difference in ASAS partial remission between treatment groups, assuming a 15% withdrawal rate.
The intention-to-treat (ITT) population was used for efficacy analyses and included all patients who were randomised, received at least one dose of study medication and had at least one efficacy evaluation after baseline. Analyses included observed data. For the primary efficacy analysis, patients who withdrew before week 28 were categorised as not achieving partial remission.
Treatment group differences in categorical efficacy measures were analysed with Fisher exact tests at a two-sided significance level of 0.05. Treatment group differences in continuous measures were analysed with analysis of covariance, with baseline values as covariates.
The safety population included all patients who received at least one dose of study medication. Adverse events were analysed descriptively.
Results
Patient disposition
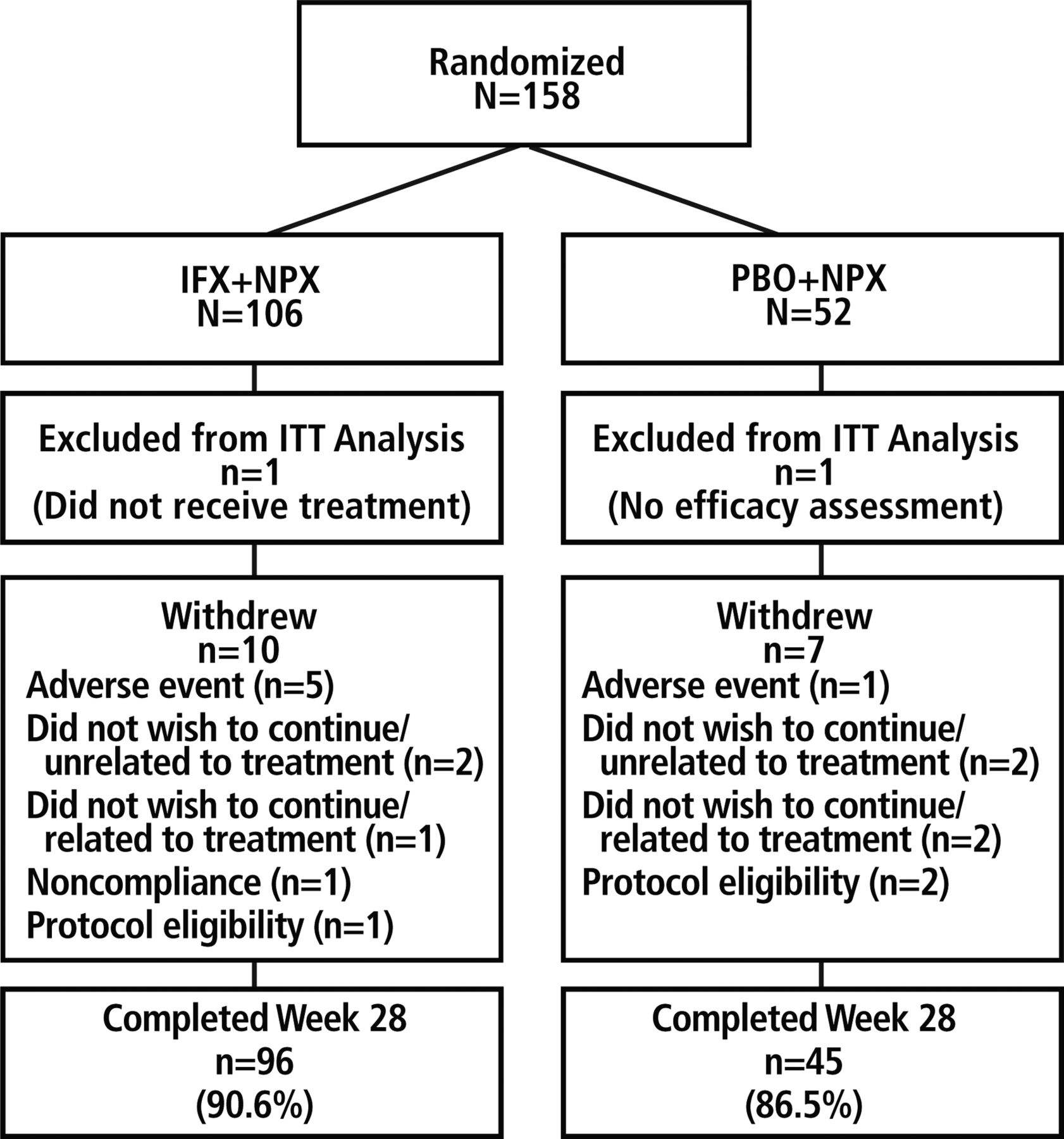
Of the 158 randomised patients, 106 were assigned to IFX+NPX and 52 to PBO+NPX (figure 1). The ITT population included 105 patients in the IFX+NPX group (one patient did not receive study medication) and 51 in the PBO+NPX group (one patient had no postbaseline efficacy assessment). The majority of patients completed the study through week 28 (90.6% in the IFX+NPX group and 86.5% in the PBO+NPX group).
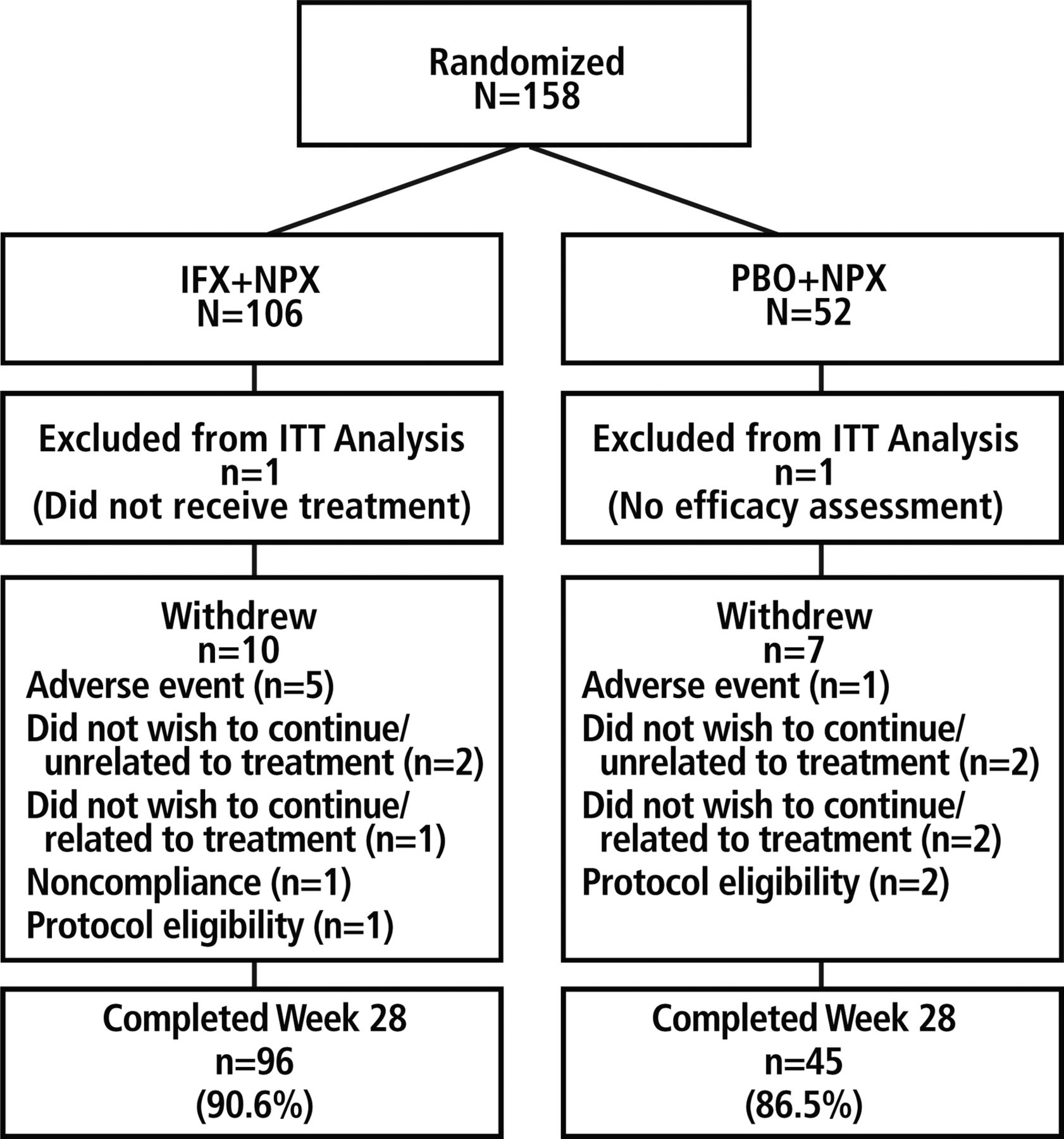
Patient disposition. IFX, infliximab; ITT, intention-to-treat; NPX, naproxen; PBO, placebo.
Baseline characteristics
Baseline characteristics were similar in the two treatment groups (table 1). Most patients had high or very high disease activity, as measured by Ankylosing Spondylitis Disease Activity Score (ASDAS), and mean time since axial SpA diagnosis was less than 1 year. The mean number of baseline SpA manifestations was comparable between treatment groups (3.8 vs 4.0). The incidence of arthritis appeared to be greater for the IFX+NPX arm than the PBO+NPX arm (45.3% vs 26.9%, respectively), but in both groups few joints were swollen (means, 1.49 vs 0.78, respectively) or tender (means 4.06 vs 3.80, respectively). Approximately 60% of patients had x-ray findings that met the modified New York radiographic criteria for AS (bilateral ≥grade 2 or unilateral ≥grade 3) at baseline, according to the x-ray reading by the local investigator.
Baseline demographic and disease characteristics
Exposure and adherence
Most patients received all six infusions of IFX or PBO (90.5% and 88.5% of patients, respectively). The mean doses per infusion of IFX and PBO were 367 and 372 mg, respectively. Treatment adherence with NPX, based on total number of doses taken regardless of dosage amount, was a mean of 99.0% for IFX+NPX and 99.5% for PBO+NPX; the mean daily doses were 960.5 and 978.1 mg, respectively.
Efficacy results
The primary endpoint was met. A greater percentage of patients achieved ASAS partial remission in the IFX+NPX group (61.9% [65/105], 95% CI 52.4% to 70.6%) than in the PBO+NPX group (35.3% [18/51], 95% CI 23.6% to 49.0%; p=0.002) at week 28 (figure 2A). The greater partial remission in the IFX+NPX group than the PBO+NPX group was statistically significant as early as week 2 and at each visit until week 28. The number of patients with partial remission increased steadily in both treatment groups over the 28 weeks. A similar pattern of treatment group differences occurred with the percentage of patients achieving 40% response in ASAS criteria (ASAS-40) (figure 2B). The percentage of patients who achieved ASAS-20 was numerically greater in the IFX+NPX group than the PBO+NPX group, but the treatment group differences were smaller and not statistically significant after week 2 (figure 2C).

Percentage of patients who achieved ASAS partial remission (A), ASAS-40 response (B), ASAS-20 response (C), ASDAS major improvement (D), ASDAS clinically important improvement (E) and ASDAS inactive disease (F). p Values are from analysis of treatment group differences in change from baseline at each visit. ASAS, Assessment of SpondyloArthritis international Society; ASDAS, Ankylosing Spondylitis Disease Activity Score; CRP, C-reactive protein; IFX, infliximab; NPX, naproxen; PBO, placebo. aASAS partial remission at week 28 was the primary endpoint of the study. Subjects who withdrew prior to week 28 were not considered to be in partial remission. Patients who were missing more than two ASAS components at week 28 were considered not in partial remission. If a patient had data for at least two ASAS domains at week 28, missing data for the remaining ASAS domains were imputed using a last-observation-carried-forward approach.
ASDAS major improvement (≥2.0-point improvement from baseline) and ASDAS clinically important improvement (≥1.1-point improvement from baseline) also showed a pattern of greater improvement in the IFX+NPX group than the PBO+NPX group at each visit (figure 2D and E). The percentage of patients with ASDAS-inactive disease (ASDAS-C <1.3, using C-reactive protein (CRP) in the calculation) in the IFX+NPX group (51.4%) was much greater than in the PBO+IFX group (19.6%) at week 28 (p<0.001) (figure 2F).
As shown in table 2, the IFX+NPX group had significantly greater improvement than the PBO+NPX group in most other measures of disease activity, clinical signs and symptoms and patient-reported outcomes at almost all visits. For all of the patient-reported outcomes, including quality of life and assessments of pain, both treatment groups improved substantially after baseline, with the IFX+NPX group showing significantly greater improvement at week 28 on all but one of the eight measures.
Secondary efficacy outcomes from baseline to week 28
Safety
Overall, IFX was well tolerated, and the pattern of AEs in the IFX+NPX group was similar to that reported previously for TNF antagonists in comparable populations (table 3). The AEs that occurred in ≥5% of patients in either the IFX+NPX (N=105) or PBO+NPX (N=52) group were nasopharyngitis (10.5% and 7.7%, respectively), upper abdominal pain (7.6% and 1.9%, respectively), headache (6.7% and 3.8%, respectively) and dyspepsia (2.9% and 5.8%, respectively).
No deaths occurred. Serious AEs were reported in five patients (4.8%) in the IFX+NPX group and three patients (5.8%) in the PBO+NPX group. In the IFX+NPX group, one patient experienced chest discomfort, dizziness and dyspnea; one patient had increased hepatic enzymes (alkaline phosphatase 2 times the upper limit of normal (ULN) and gamma glutamyl transpeptidase elevated greater than 10 times the ULN; at a follow-up 7 months after study discontinuation, liver enzymes were within normal range with a slight increase in GGT); one patient had breast cancer (diagnosed 6 months after start of treatment); and one patient had pneumonia and tuberculosis (patient was from the Russian Federation and had a negative tuberculin test and normal chest x-ray at screening; hospitalised for tuberculosis 5 months after the start of treatment). In addition, one case of fetal distress syndrome and uterine hypotonus occurred in a patient in the IFX+NPX group who reported pregnancy and was discontinued from the study; the baby was reported as born healthy after caesarean section. In the PBO+NPX group, one patient had anaemia and ovarian cyst rupture, one patient had worsening of AS and one patient had atopic dermatitis.
AEs leading to withdrawal from the study occurred in four patients (3.8%) in the IFX+NPX group and one patient (1.9%) in the PBO+NPX group (see details in table 3).
Patients with treatment-emergent adverse events (AEs)
Treatment-emergent infections and infestations occurred in 27/105 patients (25.7%) in the IFX+NPX group and 9/52 patients (17.3%) in the PBO+NPX group (see details in table 3). In both treatment groups, 1.9% of patients had an increase in hepatic enzymes, including the one serious event already mentioned above in the IFX+NPX group.
Discussion
This is the first study comparing the efficacy of a combination of a TNF antagonist and an NSAID versus an NSAID alone in patients with active axial SpA who are not refractory to NSAID therapy. Nearly two-thirds of patients who received IFX+NPX combination treatment achieved ASAS partial remission at week 28, compared with about one-third of the group who received NPX alone. This pattern of greater improvement in the IFX+NPX group was apparent as early as week 2; and continued, steady improvement occurred up to week 28.
The good results in the group receiving NSAIDs alone (35% partial remission) were rather surprising. NPX and other NSAIDs have been shown to be similarly effective in AS patients,11–14 and most of these NSAID trials used a flare design similar to the design used in INFAST (ie, patients using NSAIDs before inclusion must stop treatment and demonstrate worsening symptoms to be included in the study). However, none of these trials found ASAS partial remission rates greater than about 15%.11–13 Although the controlled phase of these studies was usually shorter (12 weeks rather than 28 weeks), response rates did not increase during trials with longer, open-label periods.12 One possible reason for the good response in the PBO+NPX group in the INFAST study may be the short symptom duration required for study entry (≤3 years, with actual mean duration <2 years) and, therefore, a mixture of patients with AS and patients with nonradiographic axial SpA; other studies have used populations with longstanding AS. In addition, all patients in INFAST had lesions seen on MRI at study entry, which was not a selection criterion for the NSAID studies. Although placebo response in the PBO+NPX group cannot be excluded, another study of patients with active axial SpA (refractory to previous NSAID therapy) with a symptom duration of <3 years found a placebo response rate of only 12.5%.6 Our data suggest that axial SpA patients respond better to a full NSAID dose if they are treated early.
Within the first 2 weeks of NSAID therapy for patients in the PBO+NPX group, a strong improvement in disease activity was seen; mean BASDAI decreased from 6.3 to 4.4, with a continued, slower decrease up to week 28 (mean, 3.2). These data suggest that response to NSAIDs can be judged in the first 2–4 weeks of treatment, as noted in the ASAS treatment guidelines,3 and that further improvement may occur with continued treatment. Similar to the results for BASDAI, a reduction of about 50% from baseline was seen in the other continuous outcomes (figure 3; table 2), including CRP concentration, which has previously been shown to improve during NSAID treatment in patients with AS.13 ,14

{kind=link}
{kind=link}
{kind=link}
Efficacy measures from baseline to week 28: BASDAI (A), BASFI (B), and ASDAS (C). p Values are from analysis of treatment group differences in change from baseline at each visit. ASDAS, Ankylosing Spondylitis Disease Activity Score; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; BASFI, Bath Ankylosing Spondylitis Functional Index; IFX, infliximab; NPX, naproxen; PBO, placebo; VAS, visual analogue scale.
Despite the better-than-expected response in the PBO+NPX group, a greater improvement in disease activity was observed in those patients who received IFX+NPX. The superiority of treatment with IFX+NPX was most obvious in the binary outcome parameters, which measured low disease activity status or major response rates (eg, ASAS partial remission and ASDAS inactive disease rate, followed by ASAS-40 response). Interestingly, there was no clear difference between the treatment groups for minor response levels such as ASAS-20. For nearly all the continuous efficacy measures, approximately 75% reductions from baseline were seen in the IFX+NPX group, and approximately 50% reductions were seen in the NPX-alone group. Whether a combination of IFX+NSAID would be superior to IFX alone cannot be determined from the INFAST study because no IFX-alone group was included. However, information about a potentially additive effect of NSAIDs and TNF antagonists for improvement of signs and symptoms would be of interest, especially given the reported inhibitory effect of NSAIDs on radiographic progression in AS (eg, refs. 15, 16).
The response rates in the IFX+NPX group of INFAST are comparable to those from a smaller, placebo-controlled IFX trial in active axial SpA patients with symptom duration of <3 years6: ASAS partial remission rates were 61.9% vs 55.6%, and ASAS-40 response rates were 67.6% vs 61.1% in the two trials, respectively. No detailed information was given in the smaller trial on the status of NSAID treatment, but these patients are likely to have been NSAID failures. The comparable response rates in the two trials and the good response rate to NSAIDs in the INFAST study support an early step-up treatment approach, with early diagnosis and treatment with NSAIDs, escalating to combination treatment after an insufficient response,3 although such an approach was not specifically tested in our trial. Currently, these two IFX studies are the only ones that limited symptom duration of axial SpA to 3 years and included a blinded control group. In a recent trial of etanercept in axial SpA, symptom duration at inclusion was limited to 5 years, and the two treatment groups, etanercept versus sulfasalazine, were not blinded for the clinical outcome assessments.7 At week 48, ASAS partial remission was reached in 50% of the etanercept group versus 19% of the sulfasalazine group. In two trials of adalimumab in patients with nonradiographic axial SpA with no limit on symptom duration, a better response rate was reported in a subgroup of patients who had shorter symptom duration.5 ,17
Fulfillment of the ASAS criteria for axial SpA and positive MRI findings of the SI joint were, according to the ASAS definition, mandatory for inclusion into the INFAST trial. Local investigators judged whether patients had radiographic sacroiliitis according to the modified New York criteria, but these judgments were irrelevant for inclusion in the study. The percentage of patients with radiographic sacroiliitis in INFAST (59%), based on the local reading of the x-rays, was relatively high compared to the other IFX trial described above that had a similar symptom duration (12%6), but the percentage was not so different from other analyses of patients with axial SpA in the first 3–5 years of their disease (50%7 ,18). Some of the variability across studies may be due to the challenge of reading x-rays of patients with early sacroiliitis; rather low sensitivity and specificity have been reported in some studies.19 ,20
Overall, the current study results, combined with evidence from other studies in which patients were already optimised on NSAID treatment, suggest that response to anti-TNF therapy may be improved with selection of patients who are young, have short disease duration and have objective evidence of inflammation (as demonstrated by MRI and elevated CRP). Evaluation of the best strategy for maintenance and/or continued improvement in patients who had achieved partial remission after 28 weeks of therapy with either IFX+NPX or NPX alone was the subject of a follow-up phase of the INFAST study that continued to week 52. Predictors of maintenance of remission were evaluated in that study, and the data will be reported in a separate manuscript.
The AE profiles for each treatment arm were as expected in patients receiving TNF-α–targeted therapy and NSAID therapy. Both treatments appeared to be well tolerated, and no new safety signals were identified. Good tolerability appears to have been reflected in the drug adherence rates, which were close to 100% in both treatment groups. Safety and good drug tolerability are especially important in this young patient population in the early phase of their disease.
In conclusion, results of the INFAST study demonstrated better outcomes on a variety of efficacy measures in patients with early axial SpA who were treated with IFX+NPX than in those treated with NPX alone. Overall, available evidence supports early diagnosis and treatment of SpA with a full dose of NSAIDs first, escalating to combination NSAID+TNF antagonist treatment in patients who have insufficient response.
Acknowledgments
Medical writing assistance was provided by Ellen Stoltzfus, PhD, and Sarah Thornburg, MS, of JK Associates, Inc., Conshohocken, PA. This assistance was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Whitehouse Station, NJ.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figure
Footnotes
Handling editor Tore K Kvien
-
Contributors All authors are responsible for the work described in this paper and were involved in at least one of the following: conception, design, acquisition, analysis, statistical analysis and interpretation of data. All authors were involved in drafting the manuscript and/or reviewing the manuscript for important intellectual content. All authors provided final approval of the final version.
-
Funding Financial support for this study was provided by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Whitehouse Station, NJ.
-
Competing interests J Sieper: Grant/Research support from Abbott, Merck, Pfizer, Janssen; Consultant for Abbott, Merck, Pfizer, UCB, Roche, Lilly, Novartis; Speakers Bureau for Merck, Abbott, Pfizer, UCB. J Lenaerts: Honoraria fees from Abbott, BMS, MSD, Pfizer, Roche, Astra Zeneca. J Wollenhaupt: Honoraria, speaker fees, or consultancy payments from a commercial entity that could potentially benefit from the publication of the subject matter discussed in the manuscript. M Rudwaleit: Consultant and Speakers Bureau for Abbott, BMS, MSD, Pfizer, UCB; Speakers Bureau for Roche. V Mazurov: None declared. L Myasoutova: None declared. S-H Park: None declared. Y-W Song: None declared. R Yao: Employee of Merck. D Chitkara: Employee of Merck. N Vastesaeger: Employee of Merck.
-
Ethics approval Appropriate local review boards (multiple).
-
Provenance and peer review Not commissioned; externally peer reviewed.