Article Text

Abstract
Objective To determine the prevalence of upregulation of interferon (IFN) type I inducible genes, the so called ‘IFN type I signature’, in CD14 monocytes in 69 patients with primary Sjögren's syndrome (pSS) and 44 healthy controls (HC) and correlate it with disease manifestations and expression of B cell activating factor (BAFF).
Methods Expression of IFI44L, IFI44, IFIT3, LY6E and MX1 was measured using real time quantitative PCR in monocytes. Expression values were used to calculate IFN type I scores for each subject. pSS patients positive for the IFN type I signature (IFN score≥10) and patients negative for the signature (IFN score<10) were then compared for clinical disease manifestations and BAFF expression. A bioassay using a monocytic cell line was performed to study whether BAFF mRNA expression was inducible by IFN type I activity in serum of patients with pSS.
Results An IFN type I signature was present in 55% of patients with pSS compared with 4.5% of HC. Patients with the IFN type I signature showed: (a) higher EULAR Sjögren's Syndrome Disease Activity Index scores; higher anti-Ro52, anti-Ro60 and anti-La autoantibodies; higher rheumatoid factor; higher serum IgG; lower C3, lower absolute lymphocyte and neutrophil counts; (b)higher BAFF gene expression in monocytes. In addition, serum of signature-positive patients induced BAFF gene expression in monocytes.
Conclusions The monocyte IFN type I signature identifies a subgroup of patients with pSS with a higher clinical disease activity together with higher BAFF mRNA expression. Such patients might benefit from treatment blocking IFN type I production or activity.
This is an open-access article distributed under the terms of the Creative Commons Attribution Non-commercial License, which permits use, distribution, and reproduction in any medium, provided the original work is properly cited, the use is non commercial and is otherwise in compliance with the license. See: http://creativecommons.org/licenses/by-nc/3.0/ and http://creativecommons.org/licenses/by-nc/3.0/legalcode
Statistics from Altmetric.com
Introduction
Primary Sjögren's syndrome (pSS) is an autoimmune disease characterised by lymphocytic infiltrates in salivary and lachrymal glands. After rheumatoid arthritis, pSS is the second most common generalised autoimmune disease.1 Nevertheless, establishment of the diagnosis is difficult owing to heterogeneity of the disease and lack of a specific diagnostic test. The diagnosis of pSS is based on 2002 American–European classification criteria.2 Treatment is mainly symptomatic and the efficacy differs across patients. If new biomarkers based upon underlying pathogenic pathways can be identified, then more effective, evidence-based treatments for pSS might be developed.
Previously, we were the first to describe a systemic upregulation of interferon (IFN) type I inducible genes in CD14 monocytes of patients with pSS.3 This was in line with the described local increased activation of IFN type I in salivary glands of patients with pSS4–6 and confirmed in peripheral blood mononuclear cells (PBMCs).7 IFN type I has an important role in the innate immunity by inhibiting viral replication, activating natural killer cells, boosting generation and activation of dendritic cells and enhancing antibody responses.8–12 Given that there are 17 different IFN type I subtypes, however, it is difficult to measure protein levels using an ELISA. For diseases such as hepatitis C, IFN type I is part of the conventional treatment. Interestingly, development of Sjögren-like symptoms has been described upon treatment with IFN type I in patients with hepatitis C13–15 supporting the role for IFN type I in the pathogenesis of pSS.
Another factor known to be involved in the pathogenesis of pSS is B cell activating factor (BAFF) of the tumour necrosis factor family. An increased expression of BAFF, which correlates with autoantibody level, in both serum16 ,17 and salivary glands,18 has been described for pSS.16 BAFF-transgenic mice develop Sjögren-like symptoms.19 IFN type I has been shown to induce BAFF expression in cultured monocytes and salivary gland epithelial cells of patients with pSS20 ,21 and a correlation between IFN type I and BAFF has been shown in patients with multiple sclerosis treated with IFN type I.22 BAFF dependence of IFN type I functioning mechanisms has been observed in systemic lupus erythematosus (SLE)-prone mice.23
Previously, we found a significant upregulation of 23 IFN type I inducible genes using whole genome analysis of pooled monocyte samples of patients with pSS. Further assessment of the prevalence of the monocyte IFN type I inducible gene overexpression in pSS and correlations with disease manifestations have yet to be performed. In this study, we therefore first measured the expression of 11 IFN type I inducible genes, which were previously detected by us, in CD14 monocytes from 69 patients with pSS and 44 healthy controls (HC). Results of factor analysis showed that five genes (IFI44, IFI44L, IFIT3, LY6E and MX1) explained 95% of the total variance of the 11 genes, and we therefore decided to adopt overexpression of these five genes as our operational definition of positivity for an ‘IFN type I signature’. The relationship between this IFN type I signature in monocytes and various disease manifestations was then studied. Given the data suggesting that BAFF is an IFN type I inducible factor, we further decided to measure the in vivo BAFF mRNA expression in monocytes of patients with pSS and to correlate the expression level with the IFN type I signature.
Patients and methods
Patients
Sixty-nine patients with a positive diagnosis for pSS according to 2002 American–European criteria2 were recruited. Patients treated with prednisone >10 mg daily, immunosuppressant or biological agents were excluded. The level of disease activity was assessed using EULAR Sjögren's Syndrome Disease Activity Index (ESSDAI).24 Forty-four HC with no autoimmune diseases and not using corticosteroids were included. Characteristics of patients and controls are summarised in table 1. The medical ethical review committee of the Erasmus MC approved the study and written informed consent was obtained.
Demographics, laboratory and clinical characteristics of participants
Blood collection and isolation of monocytes
Blood was collected in clotting tubes for serum preparation (stored at −80°C) and in sodium-heparin tubes for PBMC preparation, as described previously.25 CD14-positive monocytes were isolated as described.25
Real-time quantitative-PCR (RQ-PCR)
Total RNA was isolated from purified monocytes followed by cDNA preparation and RQ-PCR analysis using predesigned primer/probe sets (Applied Biosystems, Foster City, California, USA).25 For calculation of relative expression, all samples were normalised against expression of the household gene Abl.26 Fold change values were determined from normalised CT values using 2−ΔΔCT method (User Bulletin, Applied Biosystems).
Measurement of complement, immunoglobulin levels and autoantibodies
C3 and C4 were measured using Immage nephelometer (Beckman Coulter, Woerden, The Netherlands).
IgG, IgA, IgM were measured by turbidimetry using an Modular P800 (Roche, Almere, The Netherlands).
Anti-SSA and anti-SSB were determined by EliA (Thermo Scientific, Uppsala, Sweden) and confirmed with ANA profile immunoblot (EuroImmun, Lubeck, Germany). When the results showed a discrepancy, a QUANTA Lite ELISA kit from INOVA (San Diego, USA) was used for confirmation.
Bioassay for BAFF activity in serum and ELISA
To assess whether BAFF mRNA expression could be induced by IFN type I activity in the serum of patients with pSS, THP-1 cells were cultured in 250 Μl medium, as described previously.3 Serum (250 Μl) from patients with pSS positive or negative for the IFN type I signature was added to THP-1 cells and incubated for 6 h. As positive control, recombinant human IFN-α (symbol alpha instead of ‘A’) was added (5 ng/ml, Peprotech, London, UK). HC serum was added as negative control and for blocking anti-IFN type I receptor (PBL Interferon Source, Piscataway, USA) was added (5 Μg/ml).
BAFF protein in serum was assessed by ELISA (improved Quantikine Human BAFF/BLyS, R&D Systems, Minneapolis, USA).
Factor analysis
Expression levels of 11 IFN type I inducible genes were submitted to a principal component analysis to identify correlated groups of genes and reduce data complexity. The Kaiser–Meyer–Olkin measure of sampling adequacy was 0.833 with significant Bartlett's test of sphericity (p<0.001). Eigenvalues were derived to assess the amount of variance explained by each component factor.
Statistical analyses
Statistical analyses were performed using SPSS 17.0 package. When data were not normally distributed, values were expressed as medians with IQRs and comparisons were made using the non-parametric Mann–Whitney U test. For normally distributed data, independent t test was used to compare means. Correlations were assessed using either Pearson correlation test for normally distributed data or Spearman's Ρ when data were not normally distributed. Linear regression was performed on ESSDAI components with the IFN score as the dependent variable. Differences were considered statistically significant if p<0.05.
Results
Increased IFN type I inducible gene expression in pSS monocytes
In whole genome analyses of pooled monocytes, we previously showed that 23 IFN type I inducible genes were upregulated in pSS relative to HC.3 On the basis of their high, intermediate or low levels of upregulation in the whole genome analysis, 11 of the 23 IFN type I inducible genes were selected for analysis in this study using RQ-PCR in CD14 monocytes and found to be significantly upregulated in the pSS group relative to the HC group (figure 1A). The 11 IFN type I inducible genes analysed in the monocytes were: IFI27, IFI44L, IFIT3, IFITM1, SERPING1, IFIT1, IFIT2, LY6E, IFI44, XAF1 and MX1.

(A) Gene expression of 11 interferon (IFN) type I inducible genes in patients with primary Sjögren's syndrome (pSS) (n=69) and healthy controls (HC) (n=44). To compare means the Mann–Whitney U test was applied. (B) Heat map showing gene expression of five interferon (IFN) type I inducible genes in monocytes of patients with pSS (n=69) and HC (n=44). On the left the patients with pSS are depicted and subdivided into IFN type I signature-positive patients and IFN type I signature-negative patients. On the right the HC are depicted. Red colour indicates high gene expression. (C) Distribution of IFN scores in IFN type I signature-positive and -negative patients and HC. IFN type I-positive cases are depicted in red. Blue lines depict medians. (D) Heat map showing gene expression of IFN type I inducible genes in monocytes of patients with pSS (n=24) at two different time points (average period between two measurements 3.6±2.5 years). (E) No significant differences detected between two time points using the dependent t test.
Prevalence of ‘IFN type I signature’ in pSS monocytes
The results of a principal component analysis showed that a subset of five genes (IFI44, IFI44L, IFIT3, LY6E and MX1) explained 95% of the total variance of the 11 IFN type I inducible genes. Given that the expression of these five IFN type I inducible genes was not normally distributed, log transformations of expression values were performed and IFN scores were calculated as described for SLE.27 The mean and SD level of each IFN inducible gene in the HC group were used to standardise expression levels of each gene for each study subject. The standardised expression levels were subsequently summed for each patient to provide an IFN type I expression score.
The distribution of the IFN scores for the 69 patients was bimodal with an overlap at a score of 10. We therefore set the threshold for a positive IFN type I signature at 10. Adoption of this threshold showed that 55% of pSS group had an IFN type I signature and only 4.5% of the HC group (figure 1B,C).
To determine if the IFN type I signature possibly changes over time, we assessed IFN scores at two different time points in 24 patients with pSS. The average period between the two measurement points was 3.6±2.5 years. A significant difference in the scores over time was not detected (figure 1C,D), which shows a stability of the signature over time.
Correlation of IFN type I signature with disease parameters
To investigate whether IFN type I activation, as reflected by a high IFN score, is associated with disease activity, we assessed ESSDAI disease activity scores in 38 patients with pSS. Significant positive correlation was observed between IFN type I scores and ESSDAI scores (r=0.458, p=0.003). The patients with pSS were next stratified according to their IFN type I signature status (IFN score <10 vs IFN score ≥10) and the disease activity scores were compared. Patients with a positive IFN type I signature (IFN score >10) showed significantly higher ESSDAI scores than those with a negative signature (figure 2A). The high disease activity for patients with a positive IFN type I signature was mostly attributable to the presence of glandular, cutaneous and haematological manifestations (table 2). Linear regression analysis showed that the glandular, cutaneous and articular manifestations—despite the non-suggestive p value in the univariate analysis—were associated with high IFN type I scores (Β coefficients with 95% CI, respectively: 5.90 (1.78 to 10.01), 5.00 (1.66 to 8.35) and 7.54 (2.38 to 12.71).
Comparison of interferon (IFN) scores for patients with primary Sjögren's syndrome with or without different Sjögren's Syndrome Disease Activity Index (ESSDAI) features

(A) EULAR Sjögren's Syndrome Disease Activity Index (ESSDAI) scores in interferon (IFN) type I signature-positive and -negative patients with primary Sjögren's syndrome (pSS) (n=38). (B) IFN scores in patients with pSS positive or negative for anti-SSA (both Ro52 and Ro60) and anti-SSB (n=69). (C) Rheumatoid factor levels in IFN type I signature-positive and -negative patients with pSS (n=50). (D) IgG levels in IFN type I signature-positive and -negative patients with pSS (n=68). (E) C3 levels in IFN type I signature-positive and -negative patients with pSS (n=46). (F) Absolute lymphocyte levels in IFN type I signature-positive and -negative patients with pSS (n=42). (G) Absolute neutrophil levels in IFN type I signature-positive and -negative patients with pSS (n=41). Independent t test was used to compare means in E–G, where horizontal bars represent the means. Mann–Whitney U test was used to compare means in A–D, where horizontal bars represent the medians. In B medians and IQR are depicted; ** represents p value<0.01 and **** represents p value <0.0001.
In addition to the ESSDAI, demographic, laboratory and clinical parameters were collected. Comparison of IFN type I signature-positive patients with IFN type I signature-negative patients showed significant differences in anti-SSA autoantibodies (anti-Ro52 and anti-Ro60), anti-SSB autoantibodies, rheumatoid factor, C3, IgG, lymphocyte and neutrophil count (table 3).
Comparison of patients with primary Sjögren's syndrome with or without a positive interferon (IFN) type I signature
When patients were next stratified according to their autoantibody status (autoantibody positive vs autoantibody negative) for comparison of their IFN scores, patients with autoantibodies showed higher IFN scores than patients without autoantibodies (figure 2B). Rheumatoid factor and higher IgG levels were more often present in the IFN signature-positive patients than in IFN type I signature-negative patients (figure 2C,D). C3, lymphocytes and neutrophils were lower in IFN type I signature-positive patients with pSS (figure 2E–G). No differences were found with respect to demographic characteristics or medication status (table 3).
Correlation between BAFF expression and IFN type I signature
As already mentioned, a factor also known to be involved in the pathogenesis of pSS and found to correlate with elevated serum levels of IgG, anti-SSA and anti-SSB is BAFF.16 BAFF mRNA expression was increased in monocytes of the pSS group relative to the HC group (figure 3A). Significant positive correlation between the IFN score and BAFF mRNA expression was also thus found (figure 3B).
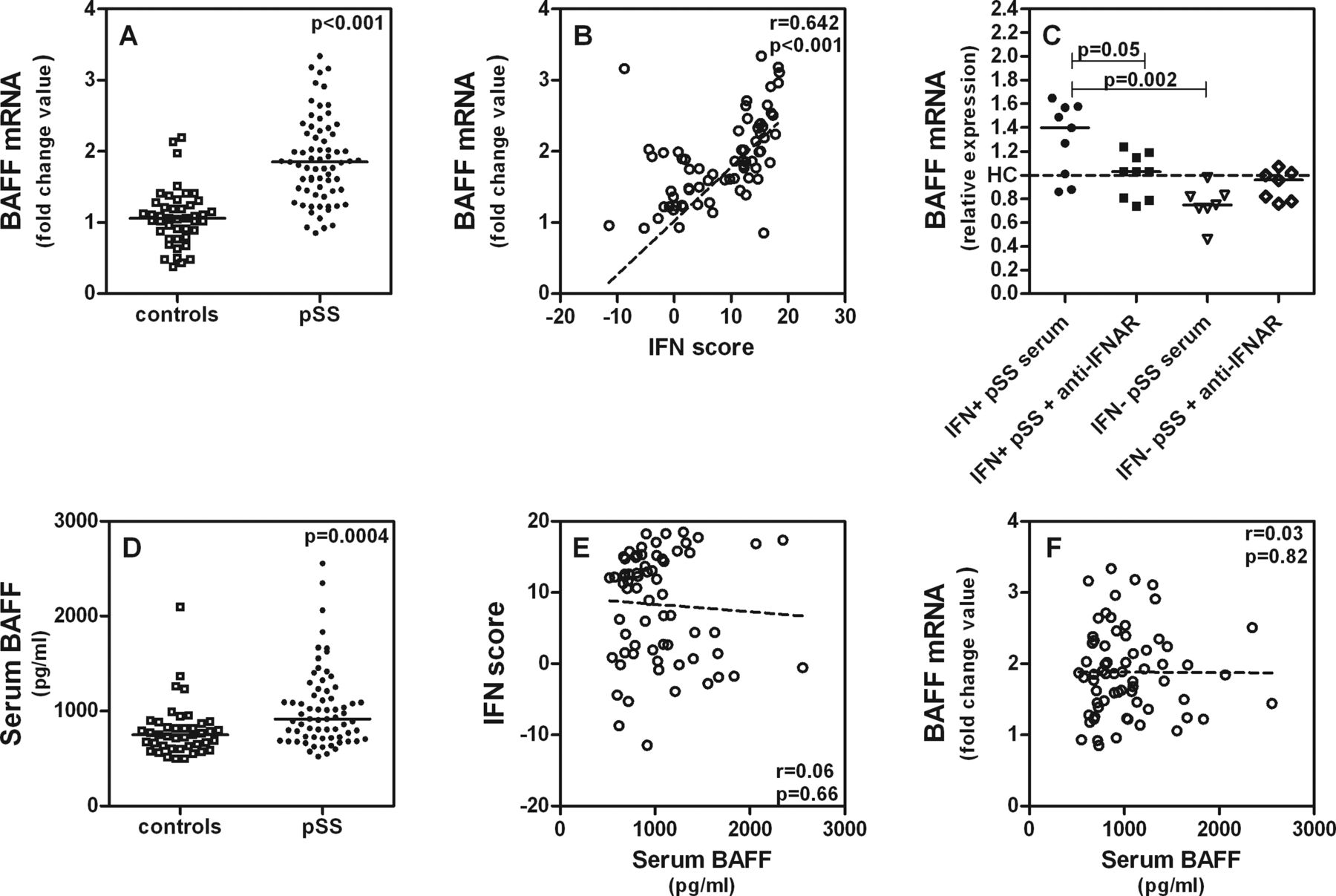
{kind=link}
{kind=link}
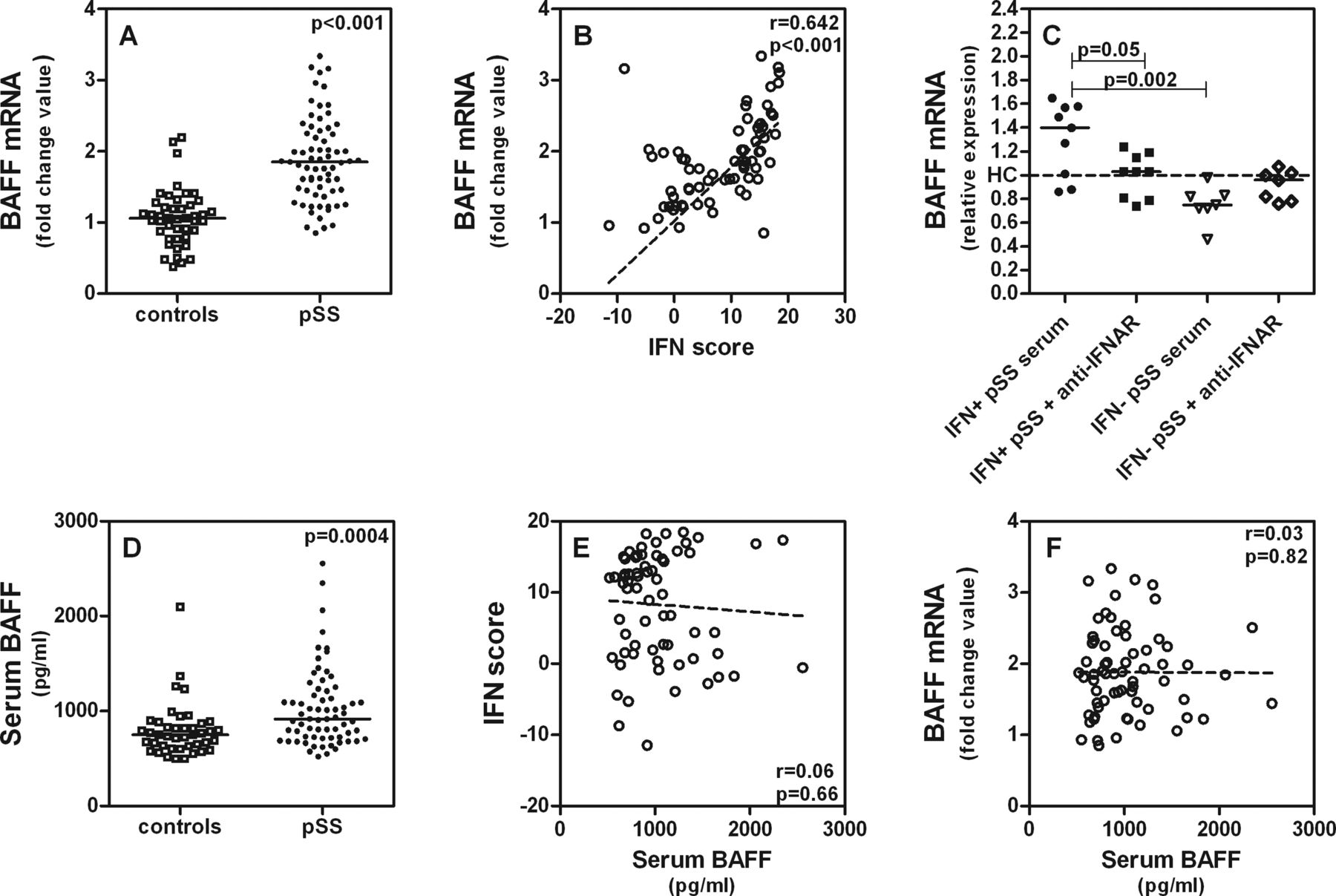{kind=link}
(A) B cell activating factor (BAFF) mRNA expression in monocytes in patients with primary Sjögren's syndrome (pSS) (n=69) and healthy controls (HC) (n=44). BAFF mRNA expression was determined by real time quantitative-PCR. (B) Correlation between interferon (IFN) type I score and BAFF mRNA expression in monocytes in patients with pSS (n=69). (C) Induction of BAFF mRNA expression in THP-1 cells by incubation with 50% serum of IFN type I signature-positive patients with pSS (n=9) and IFN type I signature-negative patients with pSS (n=7) in the presence of a blocking IFN type I receptor antibody. Expression is relative compared with the mean expression in HC (n=8). (D) BAFF protein levels in serum of patients with pSS (n=68) and HC (n=42) measured by ELISA. (E) Correlation between serum BAFF protein and IFN scores in patients with pSS (n=68). (F) Correlation between serum BAFF protein and BAFF mRNA expression in monocytes of patients with pSS (n=68). The correlation coefficients (r) and p values are shown. For correlations Spearman's ρ correlation test was used and to compare means the Mann–Whitney U test was used.
We therefore next investigated if BAFF mRNA expression could be induced in monocytes by IFN type I serum activity. A bioassay was performed for this purpose using a THP-1 monocytic cell line exposed to 50% by volume serum of patients with pSS who were either positive or negative for the IFN type I signature with and without a blocking antibody against the IFN type I receptor. After incubation with serum from IFN type I signature-positive patients (n=9), BAFF mRNA expression was indeed induced in THP-1 cells. Blocking IFN type I receptor diminished BAFF mRNA expression in THP-1 (figure 3C). Serum of IFN type I signature-negative patients did not induce higher BAFF mRNA expression relative to that found for the HC group.
To assess BAFF protein in serum, we performed an ELISA on 68 pSS samples and 42 HC. A statistical difference between pSS and HC was seen (figure 3D). After stratification for positive versus negative IFN signature, however, no differences and no correlations between the IFN scores and serum BAFF protein were found (figure 3E). It is noteworthy that serum BAFF protein does not correlate with BAFF mRNA in monocytes (figure 3F) and we also found in a smaller series that BAFF mRNA in monocytes does not correlate with BAFF mRNA in PBMCs (see supplementary online figure S1). This suggests that production of BAFF is regulated differently in different circulating leucocytes; however, this needs further investigation.
Discussion
This study shows increased IFN type I activity for 55% of a group of patients with pSS versus 4.5% for HC. The presence of such an ‘IFN type I signature’ in monocytes of patients with pSS was further shown to be associated with the ESSDAI, biological markers of activity and BAFF mRNA in monocytes. BAFF expression could also be induced in cultured monocytes by serum from patients with pSS with a positive IFN signature.
Taken together, these results suggest the following: a raised level of IFN type I, present in the serum of a significant number of patients with pSS, induces in monocytes overexpression of IFN type I inducible genes, among which is BAFF. After production, this cytokine can induce polyclonal B cell stimulation, which results in higher autoantibody production and enhanced autoantigen–autoantibody reaction with complement consumption. This scenario fits with our observation of a correlation between the presence of a positive IFN type I signature in monocytes and cutaneous manifestations in the ESSDAI index. Moreover the biological domain of the ESSDAI includes markers for B cell activation such as IgG, cryoglobulinaemia, decrease of complement. Thus part of the association between the IFN signature and the ESSDAI may be related to the role of IFN type I in B cell activation.
A similar scenario resulting in vasculitis has been described for SLE, systemic sclerosis and dermatomyositis. For subgroups of these diseases, overexpression of IFN type I induced genes has also been found. In SLE the overexpressed genes were associated—similar to the findings of our studies—with nuclear autoantibodies, glomerulonephritis and higher disease activity,28–30 again indicating the relationship between IFN activity and B cell activation and immune complex vasculitis. While these studies used PBMCs, we examined monocytes, as our focus of previous work was on the role of monocytes and dendritic cells in pSS. The IFN type I signature which we report for pSS monocytes is similar to the IFN inducible gene expression profile observed for pSS PBMCs.7 Furthermore, we assessed the IFN type I signature in PBMCs of 12 patients with pSS and six HC in supplementary analyses and found a high correlation with the IFN type I signature identified for monocytes in the same sample (see supplementary online figure S2).
There was surprisingly no correlation of monocyte activation related to an IFN type I signature with other hallmarks of pSS (ie, sicca symptoms and fatigue) (unpublished results). This is surprising as there are different reports of sicca syndrome and fatigue being induced by IFN type I treatment in patients with hepatitis C and cancer.31 ,32 All patients scored positive for the sicca syndrome and these symptoms are not quantified in the ESSDAI. Quantification using a Schirmer test or analogous test is therefore merited in future studies. For fatigue the absence of a correlation might be explained by the fact that we assessed fatigue using the Visual Analogue Scale score and not the Multidimensional Fatigue Inventory, which covers different dimensions of fatigue. There are also reports that immune activation is not the only determinant of fatigue in patients treated with IFN type I, but the induction of indoleamine 2,3-dioxygenase and the catabolism of tryptophan to quinolinic acid at the expense of serotonin are also factors.33 More in-depth research into the relations between fatigue and IFN type I signature in pSS is thus essential.
Previously, we showed that although the number of plasmacytoid dendritic cells (pDC) was lowered in the blood of patients with pSS—possibly owing to migration into the glands—the remaining pDC showed increased activation.3 There are indications that pDC are the source of the high IFN type I serum levels in SLE.34 pDC are the most powerful producers of IFN type I. Stimuli for the pDC to produce IFN type I can be either exogenous (viral DNA/RNA) or endogenous, such as immune complexes of nuclear antigens and antibodies (hallmarks of pSS) which bind to FcγRIIa on the pDC, followed by internalisation and binding to intracellular TLR7 and TLR9.35 This IFN type I production by pDC upon immune complex binding might be an explanation for the association we found between IFN type I scores and autoantibodies, IgG and lowered C3. The absence of modification of the IFN type I signature in patients treated with Plaquenil is surprising as Plaquenil is known to inhibit the activation of intracellular TLR. This might be explained by the fact that patients in our cohort are using Plaquenil for years. Assessment of IFN scores in patients before and after initiation of Plaquenil treatment in future research could therefore be informative.
At this point two limitations on our study should be mentioned. First, the age of the patient group did not completely match that of the HC group. In the statistical analysis we took that into account and could not find a significant influence of age on outcomes. A second possible limitation is the use of multiple parameters for our correlations between the presence of an IFN type I signature and clinical manifestations. Although it is questionable whether a Bonferroni correction is needed, when we applied such correction for n=22 (resulting in a p value of 0.002), the differences in rheumatoid factor, C3 and lymphocyte and neutrophil counts lost significance. To reach significance after such correction for C3, for example, 88 patients would have to be studied. Our study of 69 patients with pSS is relatively large, but new validation studies with larger numbers of participants are needed—also in light of the fact that the ESSDAI was measured in only 38 patients and laboratory parameters were not available for all patients.
To conclude, the findings of this study suggest that determining the presence of an ‘IFN type I signature’ in monocytes of patients with pSS can be used to identify subgroups of patients for specific treatment. Patients identified in such a manner may benefit from treatment aimed at blocking the IFN type I activity and thereby counteracting B cell activation and autoantibody production.
Acknowledgments
We thank Dr JJ Luime and Dr GJJM Borsboom (Erasmus MC Rotterdam, The Netherlands) for their help with the epidemiological and statistical analyses.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement - Online Figure 1
- Data supplement - Online Figure 2
Footnotes
-
Contributors ZB was involved in the study design and clinical and laboratory data collection, monitored data collection for the whole study, wrote the statistical analysis plan, cleaned and analysed the data and drafted and revised the paper. She is guarantor. NIM was involved in study design, laboratory data collection and revising the draft paper. CGvH-M was involved in laboratory data collection and revising the draft paper. JPvdM, PLvD and VAD were involved in collection of clinical data at the outpatient clinic and revising the paper. MIW was involved in collection of the laboratory data and revising the paper. WB was involved in analysing the data, drafting the figures and revising the papers. HAD was involved in study design and final draft and approval of the paper. MAV was involved in study design, monitoring of data collection, analysis of the data and final draft and approval of the paper. She is also a guarantor.
-
Funding Financial support was provided by The Netherlands Organisation for Scientific Research (NWO) and The Dutch Arthritis Association (Reumafonds).
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval The medical ethical review committee of Erasmus MC Rotterdam.
-
Provenance and peer review Not commissioned; externally peer reviewed.
-
Open Access This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 3.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/3.0/