Article Text

Statistics from Altmetric.com
The wind of change is blowing in rheumatology. Rheumatologists may soon be exposed to ‘biosimilars’ of the medicines they routinely use as the European Medicines Agency (EMA) has recently issued its final guideline on biosimilar monoclonal antibodies (mAbs)1—a product class of utmost importance for rheumatologists—and a biosimilar infliximab is currently under evaluation for marketing authorisation in Europe,2 and more will come. Rheumatologists, as it appears, are especially challenged these days as many of their patients are already successfully put on long term treatment with individualised drug regimens, including biologicals such as tumour necrosis factor α inhibitors. Apparently, introduction of ‘copy’ versions of the medicines they already know may be seen as a critical issue, although economic considerations suggest major cost savings in healthcare.3 ‘Concerns’, ‘challenges’ and ‘critical issues’: these are terms with which the term ‘biosimilar’ is often combined in papers written by clinicians and discussions at conferences.4–6 We read about the ‘complexity’ of biological medicines, and that it is difficult to replicate their exact structure. Also in clinical literature in rheumatology, we read about ‘small variations’ that can impact the activity and function of a biosimilar, and ‘major safety and efficacy concerns’ that have to be addressed before clinicians can use biosimilars.7
In their review in this issue, Dörner et al report on a roundtable discussion on the advent of biosimilars in rheumatology recently held at the Charité in Berlin (Germany).8 I note at least one aspect in this review that is worth emphasising—the paper puts scientific facts into perspective. For example, it reports about the complexities and microheterogeneity of mAbs/cepts but also puts it into perspective with the so-called ‘originator’ mAbs/cepts that are likewise complex. As a regulator, I have been deeply involved in the design of guidelines around biosimilars (including biosimilar monoclonal antibodies), and in discussions around public perception of biosimilars that I have experienced, it has been my impression that scientific facts are often discussed in isolation. For example, it is true that biologicals such as mAbs and -cepts are complex, and that small changes in their manufacturing process can have a large impact on their function (although I have also seen a few cases where larger changes had only little impact). This has often had the connotation to implicitly assume that biosimilars therefore may have an undetected ‘inferior’ quality compared with the established originators, or that at least there is more uncertainty around them. The sentence ‘biosimilar and biological reference medicines are similar but not identical’ (also used by Dörner et al) is perhaps one of the most frequently misunderstood sentences in the history of biosimilars and has almost become a mantra when raising concerns around biosimilars. It comes from a previous version of the EMA ‘Questions and answers’ document on biosimilars9 (the document has been updated so the sentence can no longer be found there), and its intention had rather been to clarify that due to inherent complexity and inherent variability of biologicals, any biosimilar can never be ‘identical’ to its reference product (and therefore the ‘generic pathway’ is not possible). One would have to add that also no batch of any reference product is ‘identical’ to the previous one—‘non-identicality’ is a normal feature of biotechnology that has to be controlled by tight specifications of critical product attributes, within current technical and scientific limitations (inherent variability). The ‘art’ for a biosimilar is to demonstrate that the biosimilar is as close as possible to its reference product in all relevant functional and structural aspects, again within current technical and scientific possibilities and its inherent variability. What is often not mentioned (but mentioned by Dörner et al; this is also acknowledged in other papers such as Scheinberg and Kay7) is that originator mAbs/cepts have undergone changes after their approval—this is what regulators call the ‘life cycle’ of a medicine. This is normal—manufacturing processes are updated during the life cycle of any medicine, and this is welcome as these are often improvements. A recent publication suggested that licensed biologics undergo changes in relevant molecular attributes over time.10 In fact, the seminal regulatory guideline ICH Q5E, issued by the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH),11 an internationally agreed standard that sets the data requirements for changes to the manufacturing process for biotechnological and biological products, stipulates that “The demonstration of comparability does not necessarily mean that the quality attributes of the pre-change and post-change product are identical, but that they are highly similar and that the existing knowledge is sufficiently predictive to ensure that any differences in quality attributes have no adverse impact upon safety or efficacy of the drug product”. This is called the ‘comparability exercise’,12 and the determination of comparability is usually based on a combination of physicochemical and analytical testing, biological assays and, in some cases, non-clinical and clinical data. In other words, clinical data (especially from new randomised controlled clinical studies) have not always been part of a dossier for a biological medicine that underwent changes in its manufacturing process because such data were in many cases not necessary to demonstrate that the pre- and post-change product are comparable. Such changes in manufacturing can be seemingly small (ie, change in the supplier of cell culture media) and range to major ones (ie, introducing new purification steps or implementing new manufacturing sites). It is true that even small changes can have a large impact. But this would certainly be picked up in the comparability exercise and regulatory assessment, and via implementation of correct regulatory measures for post-approval surveillance. In fact, all of the licensed mAbs and -cept fusion proteins used in rheumatology have had changes in their manufacturing processes after their initial approval (figure 1A).

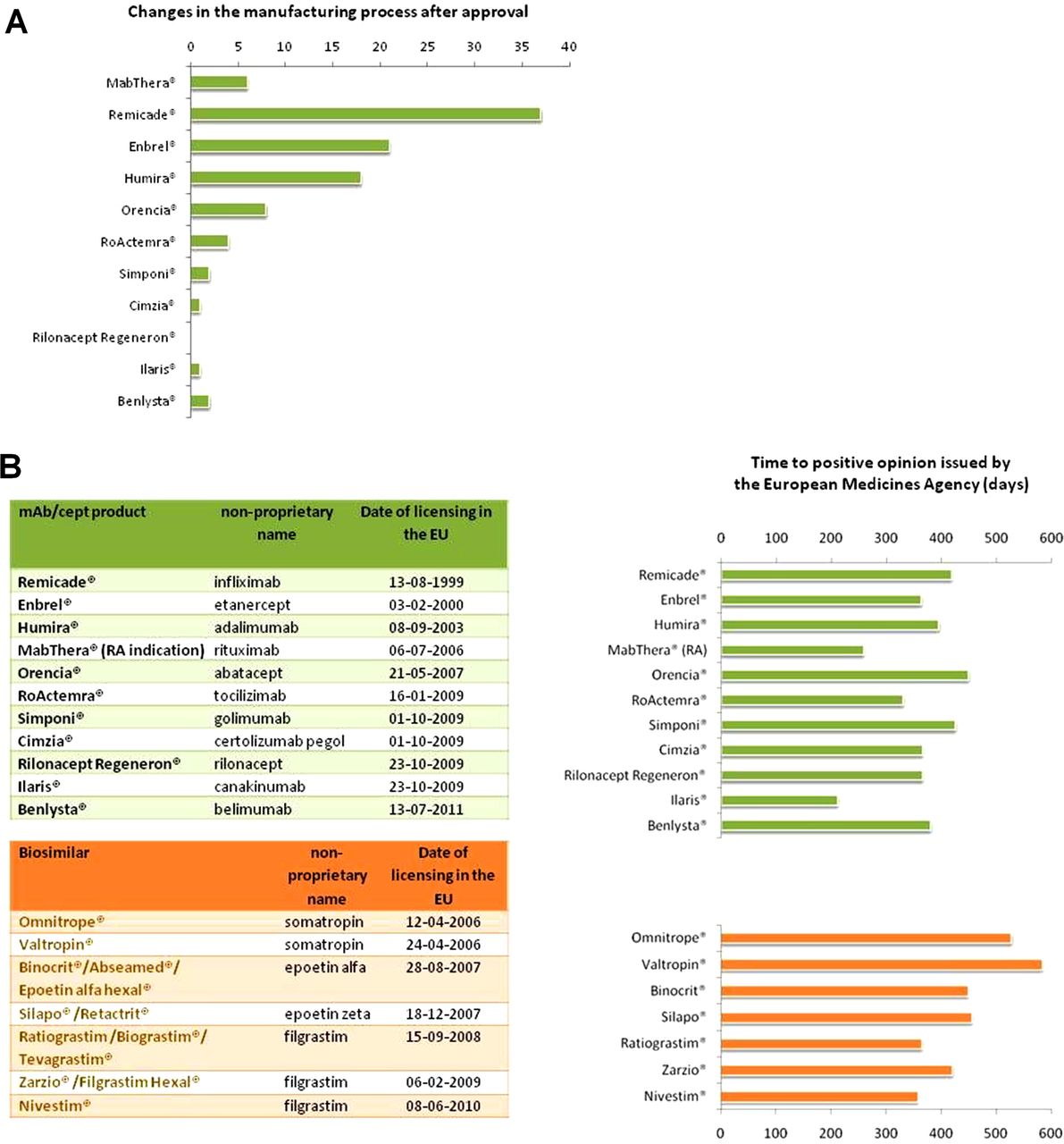{kind=link}
Number of changes in the manufacturing process after approval for monoclonal antibodies (mAbs)/cepts authorised in rheumatological indications (A). Products in order of date of approval in Europe (from MabThera, authorised on 2 June 1998 for the initial authorisation in oncology, to Benlysta, licensed on 13 July 2011). Such changes range from change in the supplier of a cell culture media to new purification methods or new manufacturing sites. (B) mAb/cept products authorised in rheumatological indications, currently licensed biosimilars (both sorted by date of licensing in the EU) (left) and time to positive opinion issued by the European Medicines Agency (EMA) (including clock stop periods where the applicants address the issues raised by EMA) (right). A positive opinion is a prerequisite for authorisation by the European Commission. All information taken and analyses made from data published in the EPAR13 (European Public Assessment Report; document type: ‘EPAR—Procedural steps taken and scientific information after authorisation’12 or ‘EPAR—Procedural steps taken before authorisation’14). NB: In (B), MabThera calculated for the rheumatoid arthritis (RA) indication, granted in 2006, not for the initial approval process in 1998 in oncology. Remicade cited for the Crohn's disease indication; RA indication granted a year later (27 June 2000).
For several years, EMA has published a regularly updated list of ‘steps taken after approval’,15 for any medicine. These can be as simple as a change in the address of the marketing authorisation holder (which obviously does not have any impact on the product) and range to complex changes such as new clinical indications or changes in the manufacturing process. One can see from figure 1A that virtually any medicine has undergone numerous changes in their manufacturing processes, and some people say that the medicine that a clinician administers to a patient today is not ‘identical’ (but comparable) to the medicine authorised years ago. Any such changes in the manufacturing process (ranging from a change in the supplier of cell culture media to new purification methods or new manufacturing sites) was substantiated with appropriate data and was approved by the EMA.
The scientific principles of a change in manufacturing process of an originator mAb/cept molecule and the generation of a biosimilar are the same.16 ,17 A ‘true’ biosimilar, as developed along the principles of the EMA guidelines, was recently proposed as a ‘copy version of an already authorised biological medicinal product with demonstrated similarity in physicochemical characteristics, efficacy and safety, based on a comprehensive comparability exercise’.17 Regulators require an as close as possible resemblance of the biosimilar to its originator, as exemplified by data recently published for some of the authorised biosimilars.18 ,19 ‘Non-innovator’ products that are copies of authorised products20 are not necessarily ‘biosimilars’, according to the current definition. The current concept of development of biosimilar mAbs/cepts follows the principle that an extensive state of the art physicochemical, analytical and functional comparison of the molecules is complemented by comparative non-clinical and clinical data that establish equivalent efficacy and safety in a clinical ‘model’ indication that is most sensitive to detect any minor differences (if these exist) between biosimilar and its reference mAb also at the clinical level.21 This ‘model’ indication has to be scientifically duly justified by the applicant, and any extrapolation to other indications not specifically studied is based on an indepth scientific review process. As such, a biosimilar development is therefore not so much ‘abridged’ but rather ‘tailored’ towards a distinct scientific objective—that is, to establish biosimilarity, not to re-establish benefit for the patient (this is also nicely described in the review by Dörner et al).
Like most biologicals, biosimilar mAbs/cepts will have to be licensed via the centralised marketing authorisation route (ie, via the EMA),22 and one could say that only on scientific assessment and positive recommendation by the EMA's Committee for Medicinal Products for Human Use (CHMP) can it be considered that the comparability between a proposed biosimilar and its reference product is established and the criteria for naming the medicine as ‘biosimilar’ have been fulfilled. The approval process for a biosimilar is critical and stringent, and no step in this process is ‘automatic’ and without scientific assessment by experts from regulatory authorities. The CHMP appoints two ‘rapporteurs’ among its members whose expert teams perform an independent critical assessment on a biosimilar candidate's quality, non-clinical and clinical data.13 In the regulatory review procedure for a marketing authorisation, dedicated experts in analytical, physicochemical and biological characterisation prepare an assessment report, and the CHMP's Biologics Working Party (BWP),23 discusses the assessment report in depth to adopt a list of questions to the applicant. The BWP is composed of European experts selected according to their specific expertise (one member per member state). An important part of the scientific assessment of a biosimilar is to put microheterogeneity (often mentioned in connection with ‘concerns’ around biosimilars) into perspective with the very same microheterogeneity found in batch to batch variability of the reference biological. As already mentioned, microheterogeneity is not specific to biosimilars; it is a ‘normal’ feature of any biological medicine. In parallel, non-clinical and clinical experts in the rapporteurs’ teams prepare separate assessment reports of the respective data and raise relevant questions in their field of expertise. The CHMP will then integrate these recommendations into a multidisciplinary list of questions for the applicant that need to be addressed before a positive recommendation for marketing authorisation can be issued. The impact of any ‘small differences’, if present, would be picked up and discussed. This holistic assessment of quality, non-clinical and clinical data puts any minor differences between the biosimilar candidate and its reference mAb into the perspective of their clinical relevance (NB: more than only ‘minor’ differences would not be in line with a biosimilar, and authorisation as a biosimilar may not be possible1).
When comparing the overall number of questions asked in these lists of questions for licensed biosimilars with those asked for mAbs/cepts authorised in rheumatological indications, one easily recognises that the review process for a biosimilar is as critical as for an originator biological. The median overall number of questions asked for currently authorised biosimilars was 119, compared with 121 for authorised mAbs/cepts used in rheumatology (analysis from confidential data on file). Obviously, these questions have been resolved following adequate responses as all of these products have subsequently been recommended for marketing authorisation. A large number of questions is not necessarily a measure of the quality of a dossier but these data show that regulatory review for biosimilars is as critical as for a new in class compound. Certainly the aforementioned figures suffer from the low number of procedures (11 biologics authorised in rheumatological indications and seven distinct biosimilar molecules), and it is scientifically not absolutely correct to compare the currently licensed biosimilars to mAbs/cepts as the biosimilars authorised to date are less complex biologics (eg, growth hormone, filgrastim or erythropoietins). However, upcoming biosimilar mAb/cept applications may not be expected to have fewer questions as these are more complex molecules, and so the conclusion that regulatory review is as critical as for originator mAbs/cepts will very likely remain valid. It can also not be said that biosimilars undergo an ‘abridged’ or ‘accelerated’ approval as the time from the start of the regulatory review procedure to a positive outcome issued by the CHMP was not shorter for the biosimilars authorised to date compared with mAbs/cepts used in rheumatology (figure 1B)—biosimilars undergo a full review, and every word in the summary of product characteristics (SmPC) that informs the prescriber has been assessed and approved, including the indications granted for a biosimilar.
Obviously, regulatory review and approval sets a high entry bar for any biological medicine, including biosimilars—this is also acknowledged by Dörner et al. Regulators evaluate biosimilars cautiously, and a biosimilar is only registered if the applicant has demonstrated in sufficient detail that the biosimilar is of good quality and equivalent in efficacy and safety to its reference medicinal product. Authorised biosimilars are as good, safe and efficacious as originator biologicals. Dörner et al8 make another valid, even vital, point that the role of biosimilars in rheumatic diseases will be determined by the confidence placed in them by rheumatologists. Closer communication and interaction between regulators (explaining their framework24) and clinicians (explaining what they need to know about it to alleviate concerns) will become increasingly important. With this in mind, the paper by Dörner et al,8 an international group of clinicians is, from my perspective, a further step along this road as it provides a balanced clinical view—is the ‘wind of change’ finally blowing in this respect too?
Numerous biosimilar mAbs/cepts are under development25 and thus a clear understanding of what a biosimilar is (and what it is not) is of critical importance. Clinicians and regulators and also other stakeholders may have to join forces to come to a balanced, objective and well informed decision making process that bases medical decisions on science, in the best interest of doctors and patients, on whose behalf we regulate.
Acknowledgments
The author wishes to thank Dr Camille Vleminckx and Dr Patrick Celis from the European Medicines Agency, and the colleagues involved in internal European Medicines Agency peer review, for helpful comments on the manuscript.
References
Footnotes
-
Disclaimer The views expressed in this article are the personal views of the author and may not be understood or quoted as being made on behalf of or reflecting the position of the European Medicines Agency or one of its committees or working parties.
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed.