Article Text

Abstract
Objectives We have previously shown that both short- and long-lived plasma cells (PCs) significantly contribute to autoantibody production in NZB/W mice as a model of lupus nephritis. The aim of this study was to determine the role of autoreactive long-lived (memory) PCs refractory to immunosuppression and B cell depletion in the pathogenesis of systemic lupus erythematosus.
Methods Splenic CD138+ antibody-secreting cells (ASCs) from >6-month-old NZB/W mice with high titres of anti-dsDNA autoantibodies or from Balb/c mice 5 days after secondary immunisation with ovalbumin (OVA) were adoptively transferred to immunodeficient Rag1−/− mice, in which the development of nephritis was investigated by measuring proteinuria. Total IgG and IgM as well as anti-dsDNA and anti-OVA antibody levels were followed up by ELISA. After 21 weeks the recipient mice were sacrificed so that PCs in spleen and bone marrow could be analysed using ELISPOT and flow cytometry and renal immunohistology performed.
Results The adoptive transfer of NZB/W and anti-OVA ASCs resulted in the continuous generation of anti-dsDNA antibodies and anti-OVA antibodies, respectively, exclusively by long-lived PCs that had homed to the spleen and bone marrow of recipient Rag1−/− mice. Rag1−/− mice generating autoantibodies including anti-dsDNA had reduced survival, proteinuria and immune complex nephritis with C1q, C3, IgG and IgM deposits 21 weeks after transfer.
Conclusions These findings demonstrate that autoantibodies exclusively secreted by long-lived (memory) PCs contribute to autoimmune pathology and should be considered as candidate targets for future therapeutic strategies.
- Autoimmunity
- B cells
- Systemic Lupus Erythematosus
- Autoantibodies
- Autoimmune Diseases
Statistics from Altmetric.com
Introduction
Autoantibodies directly or indirectly play an important role in the pathogenesis of autoimmune diseases such as systemic lupus erythematosus (SLE).1 During the last 15 years, our understanding of the biology of plasma cells (PCs) secreting antibodies or autoantibodies has markedly improved. It has become clear that antibody levels are mainly maintained by long-lived PCs.2–4 Based on these findings, we have demonstrated that short-lived plasmablasts (PBs)/PCs and long-lived PCs can contribute to autoantibody secretion.5 For this purpose, activated B cells differentiate into proliferating PBs already secreting antibodies. PBs highly expressing MHC class II molecules and the chemokine receptors CXCR3 and CXCR4 possess the capacity to migrate through the circulation along a chemokine gradient from the spleen and lymph nodes into the bone marrow (BM) and into inflamed tissues.6 These PBs are detectable in the peripheral blood only on day 6 after secondary immunisation7 or during the entire period of active disease in SLE patients.8–10 In lupus, cytokines such as interferon-α, IL-21 and BLyS/BAFF promote the expansion of autoreactive short-lived PBs and PCs.11–13 After successfully occupying a niche in the BM or inflamed tissue, PBs mature into long-lived PCs that secrete (auto)antibodies as long as they survive independently of antigen stimulation, T cell help14 and memory B cells.15 Consequently, long-lived PCs can be considered as memory cells.16 The remaining PBs that do not find a niche die due to apoptosis.6 Immunosuppressive/cytotoxic agents or B cell depletion does not affect the long-lived PC compartment.5 ,15 ,17 ,18 The reduction in autoantibody levels observed under these therapeutic conditions reflects the lack of freshly generated PBs.8
It has been assumed that long-lived PCs can secrete pathogenic autoantibodies driving inflammation. This is supported by the results of immunoablative therapy followed by autologous haematopoietic stem cell transplantation in refractory SLE. Immunoablation with antithymocyte globulin leads to the depletion of almost all lymphocytes, including long-lived PCs, providing the basis for long-term remission.19 Moreover, selective depletion of PCs in autoimmune NZB/W and MRL/lpr mice by treatment with the proteasome inhibitor bortezomib ameliorates or prevents lupus nephritis and prolongs survival.20 Nevertheless, neither immunoablation nor PC depletion using bortezomib can unambiguously reveal the contribution of long-lived PCs alone to autoantibody-mediated pathology.
This study aimed to elucidate the precise role of autoreactive long-lived PCs in the pathogenesis of SLE. Therefore, we established a humoral immune system consisting exclusively of long-lived memory PCs originating from NZB/W mice, a murine model of SLE, in Rag1−/− mice lacking B cells and PCs.21
Materials and methods
Mice and treatments
Female NZB/W F1 and Rag1−/− mice (B6.129S7-Rag1tm1Mom/J) were bred at the animal facility of the German Rheumatism Research Center Berlin (DRFZ) under defined, pathogen-free conditions. To distinguish between short-lived and long-lived PBs/PCs, we fed bromodeoxyuridine (BrdU; Sigma-Aldrich, 1 mg/mL) with 1% glucose in drinking water to the mice for 14 days before sacrifice and analysis. Subgroups of Rag1−/− mice received cyclophosphamide intraperitoneally at a dose of 30 mg/kg every 4 weeks. Additionally, 8–12-week-old Balb/c mice (Charles River) were immunised with alum-precipitated ovalbumin (OVA; Sigma-Aldrich) intraperitoneally and boosted 4 weeks after primary immunisation as described.14
Cell preparation and adoptive transfer
Single-cell suspensions from the spleens of NZB/W mice aged 24–28 weeks or from OVA-immunised Balb/c mice 5 days after boosting were filtered through a 70-µm cell strainer (BD Falcon), washed twice with phosphate-buffered saline (PBS)/0.5% bovine serum albumin (BSA), and incubated with anti-CD138–PE (clone 281–2; BD Biosciences) for 10 min. They were again washed and incubated with anti-PE MicroBeads (Miltenyi Biotec) for 15 min. Magnetic-activated cell sorting (MACS)-positive selection was then performed following the manufacturer's protocol to enrich antibody-secreting cells (ASCs). B cells were isolated from the negative fraction by using anti-B220-Cy5 (clone RA3.6B2; DRFZ) and anti-Cy5 MicroBeads (Miltenyi Biotec) according to the same MACS protocol. Next, 2–3×106 CD138+ASCs or 4–6×104 B220+B cells, corresponding to the proportion of contamination in the purified ASC fraction, were transferred to 8–12-week-old Rag1−/− mice by injection into the tail vein, and 1.2×106 ASCs derived from the spleen of OVA-immunised Balb/c mice were transferred to Rag1−/− mice.
Detection of serum antibodies
Serum was collected from recipient Rag1−/− mice at different time points after transfer to measure the levels of total IgG and IgM, IgG and IgM anti-dsDNA antibodies as well as IgG anti-OVA by ELISA. For this purpose, 96-well microtitre plates were coated with goat anti-mouse IgG or IgM (SouthernBiotech), OVA or calf thymus DNA (Sigma-Aldrich; 1 mg/mL) and incubated overnight at 4°C. DNA coating was performed after pre-coating the microtitre plates with methyl-BSA (Sigma-Aldrich; 1 mg/mL) at 37°C for 3 h. After blocking with PBS/3% BSA, sera were added and incubated overnight at 4°C followed by the addition of biotin-labelled goat anti-mouse IgM or IgG (SouthernBiotech) and ExtrAvidin-peroxidase (Sigma-Aldrich). TMB substrate (Thermo Scientific) was added for colour development. Extinction was measured at 450 nm after the reaction was stopped.
The EUROLINE ANA-Profile 3 (EUROIMMUN) line immunoassay procedure was modified so that a larger spectrum of antinuclear autoantibodies could be detected. Briefly, after the serum incubation and washing steps, the strips were incubated with biotin-labelled goat anti-mouse IgG (SouthernBiotech) followed by ExtrAvidin-alkaline phosphatase (Sigma-Aldrich) at room temperature, each for 30 min. After development, the strips were read according to the manufacturer's instructions.
Flow cytometry
PCs were detected with anti-CD138-PE (clone 281–2; BD Biosciences) for surface staining and anti-kappa Pacific Orange for intracellular staining (DRFZ clone 187.1). The intracellular staining was carried out after fixation with Cytofix/Cytoperm buffer (BD Bioscience) for 30 min. B cells were stained with anti-B220 (DRFZ clone RA3.6B2) and T cells with anti-CD3 (DRFZ clone 145-2C11). Absolute cell numbers were calculated based on population frequencies and total cell numbers per organ. Short- and long-lived PCs from the spleen, BM and kidney of recipient Rag1−/− mice were distinguished by analysing BrdU incorporation using a BrdU Flow Kit (BD Biosciences) according to the manufacturer's protocol. For analysing tracing and proliferation of transferred ASCs from NZB/W mice, the cells were incubated with 5 µM carboxyfluoresceinsuccinimidyl ester (CFSE; Invitrogen), washed twice and transferred into Rag1−/− mice as described above. The mice were sacrificed 3, 7, 14 and 30 days after transfer. Single-cell suspensions were prepared from BM (femur and tibia) as described previously. Analysis was performed using a FACSCanto II cytometer (BD Biosciences) and FlowJo software (Tree Star).
Detection of ASCs by ELISPOT
Ninety-six-well microtitre plates (Millipore) were coated with goat anti-mouse IgM or IgG for detection of total IgM and IgG ASCs, and were precoated with methyl-BSA and subsequently coated with calf thymus DNA for detection of anti-dsDNA ASCs.5 ,22 The spots were developed with NBT/BCIP (Thermo Scientific) and enumerated using an automated ELISPOT reader with ImmunoSpot software (Cellular Technology).
Assessment of nephritis
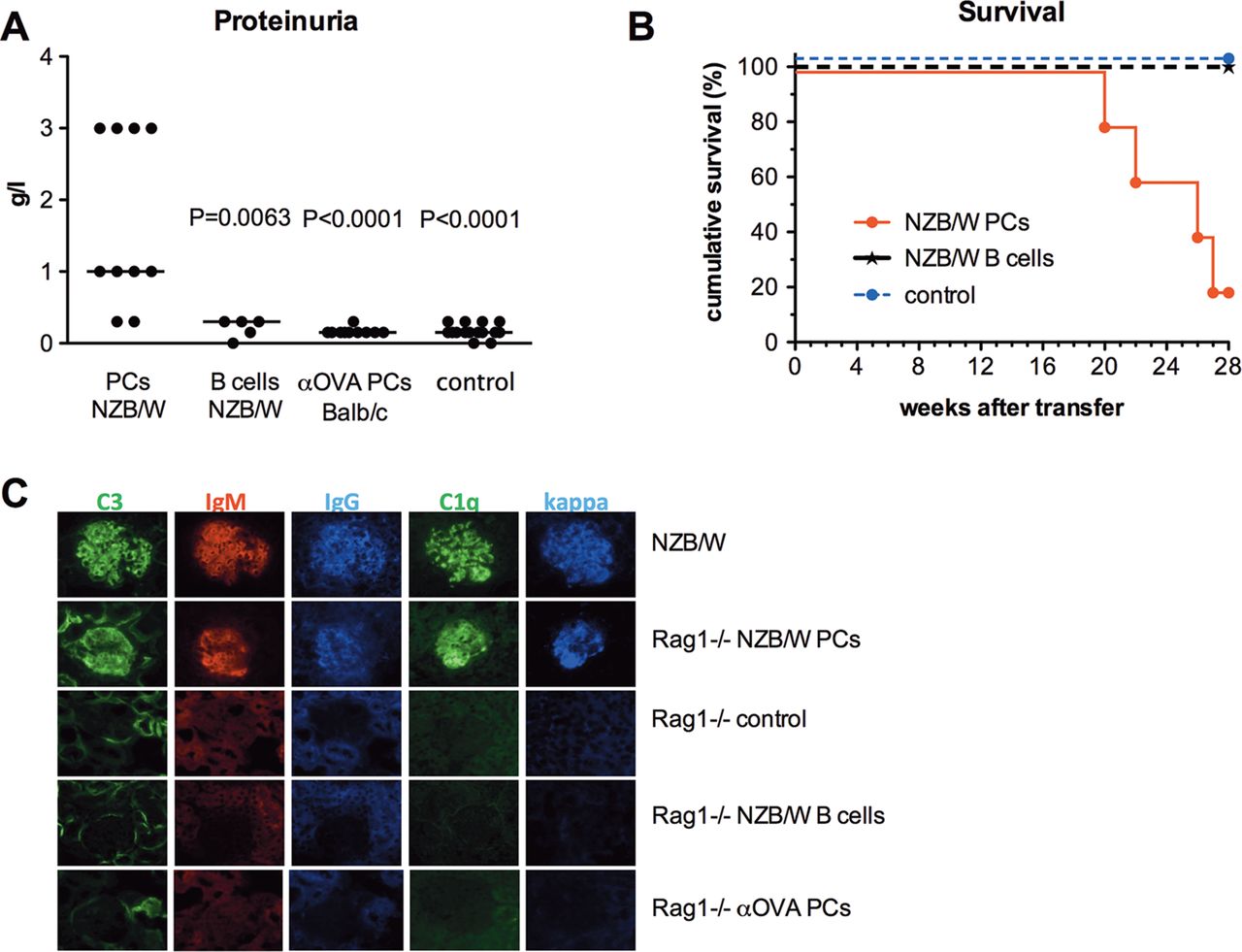
Monthly monitoring of proteinuria in recipient Rag1−/− mice was started 8 weeks after transfer using Albustix (Bayer). Immunohistology was performed on kidney cryostat sections from recipient Rag1−/− mice 21 weeks after transfer. Deposition of C1q, C3, IgM, IgG and kappa was investigated with the following anti-mouse antibodies: anti-C1q FITC, anti-C3 FITC (clones RMC7H8 and RMC11H9; Cedarlane Laboratories), anti-IgM Alexa 546 (DRFZ clone M41), anti-IgG biotin (SouthernBiotech) and anti-kappa Cy5 (DRFZ clone 187.1), respectively, followed by streptavidin APC (BD Biosciences). Slides were viewed and images were taken using a Zeiss Axioplan fluorescence microscope (Carl Zeiss).
Statistics
The non-parametric Mann-Whitney test was performed using GraphPad Prism V.5.0.
Results
Adoptively transferred PBs proliferate before they become long-lived PCs
The adoptive transfer experiments were based on the migratory capacity of PBs.6 ,8 ,23 From our previous work, we know that 60–70% of splenic ASCs from NZB/W mice belong to the compartment of short-lived PBs and PCs incorporating BrdU, indicating that these cells have recently undergone cell division. Roughly half of them are proliferating PBs since they express high levels of MHC class II.5 Thus, about 30–35% of the 2–3×106 splenic ASCs transferred into Rag1−/− mice were PBs with a migratory capacity. The purity of the CD138+ ASCs was very high, about 98% after MACS. The 2% contamination was mainly caused by B220+ B cells, while contaminating T cells made up less than 0.1% (see online supplementary figure S1). At most, 0.7–1×106 of the cells were PBs able to home to the target tissues.
In order to confirm that the transferred splenic ASCs contained proliferating and migratory PBs, the cells were labelled with CFSE before transfer and analysed in BM on days 3, 7, 14 and 30 after transfer. CD138+ ASCs were already detectable in BM 3 days after transfer (figure 1). On day 3, about 30% of the cells showed a loss of CFSE labelling intensity, indicating that they had undergone cell division in the meantime. The other cells did not proliferate within the first 3 days. Between days 3 and 14, there was a continuous increase in the number of cells showing a loss of CFSE intensity as a sign of cell division. There was no change in the CFSE intensity pattern and 28% of cells retained their full CFSE intensity from day 14 to day 30, indicating that cell proliferation was completed 14 days after transfer at the latest.

Behaviour of adoptively transferred antibody-secreting cells (ASCs) in the bone marrow (BM) of recipient Rag1−/− mice. For this analysis, ASCs isolated from the spleens of NZB/W mice were labelled with carboxyfluoresceinsuccinimidyl ester (CFSE) prior to transfer. CSFE is a membrane-permeable, fluorescein-based dye that crosslinks to intracellular proteins. We used it to track the cell division of transferred ASCs in BM in vivo as each cell division results in the progressive halving of the fluorescence intensity of the dye in cells. Histogram plots of BM ASCs gated for CD138+ and intracellular kappa+ cells in recipient Rag1−/− mice showed a loss of CFSE intensity from days 3 to 14 after transfer but no further change in CFSE intensity after that. The persistence of CSFE labelling is indicative of the absence of proliferation.
Adoptive transfer of ASCs results in exclusive survival of long-lived PCs in recipient Rag1−/− mice
CFSE-labelling of transferred splenic ASCs showed that proliferation of PBs was completed within 14 days at the latest. Afterwards, (auto)antibodies should be secreted by mature long-lived PCs surviving in niches in the BM and spleen and, possibly, other lymphoid organs and inflamed tissues.6 ,8 To confirm this, recipient mice that had received BrdU in drinking water for 2 weeks by week 21 after transfer were sacrificed and examined. Fluorescence-activated cell sorting (FACS) showed that all PCs found in the spleen, BM and kidney (figure 2A) were BrdU-negative (figure 2C). Treatment of a subgroup of Rag1−/− mice with cyclophosphamide at weeks 8, 12 and 16 after transfer did not reduce antibody levels relative to those in an untreated subgroup (figure 4A). This supports the BrdU staining data suggesting that antibodies were produced by long-lived PCs. Of note, significant B and T cell expression was not observed in the investigated organs.

Analysis of plasma cells (PCs) in the spleen, bone marrow (BM) and kidney of Rag1−/− mice 21 weeks after adoptive transfer. (A) Examples of PC analysis by fluorescence-activated cell sorting (FACS) using CD138 and intracellular kappa staining in each recipient and control Rag1−/− mouse. Numbers in the panels represent cell numbers as a percentage of the total cell number. (B) Bar graphs show the absolute numbers of PCs in the spleen, BM and kidney of recipient Rag1−/− mice as determined by FACS. Data represent the mean of 5 mice per group±SEM. (C) Histogram plots demonstrate the lack of bromodeoxyuridine (BrdU) staining in spleen, BM and kidney PCs. Panels show the number of BrdU-negative long-lived PCs as a percentage of the total PC population. (D) Absolute numbers of IgM and IgG anti-dsDNA secreting PCs in the spleen, BM and kidney of recipient Rag1−/− mice as determined by ELISPOT. Data represent the mean of 5 mice per group±SEM. OD, optical density.
PCs were mainly detected in the spleen of recipient mice 21 weeks after transfer. The number of splenic PCs was 10 times higher than in the BM, while the number of PCs in the kidney was negligible (figure 2B). Renal PC infiltrates have been observed previously in NZB/W mice with established nephritis.17 ,22 ,24 ,25 Their absence here is not surprising because the inflammatory milieu needed for homing of PCs was lacking when the cells were transferred. The FACS data show that, overall, about 2.2×105 PCs could be recovered in these organs after 21 weeks, which is 10% of the number of transferred ASCs or 25–30% of the number of estimated transferred PBs able to home to the target tissues. Although PC counts were much higher in the spleen, anti-dsDNA-secreting PCs were more abundant in the BM (figure 2D). Interestingly, the proportion of IgG anti-dsDNA secreting cells was significantly higher in the BM than in the spleen (table 1).
Percentage of anti-dsDNA-secreting cells (ELISPOT) in the total antibody-secreting cell (ASC) population in the spleen and bone marrow (BM) of NZB/W donor mice aged 24 to 28 weeks versus Rag1−/− mice 21 weeks after adoptive transfer of ASCs from NZB/W mice
Long-lived (memory) PCs continuously secrete (auto)antibodies in recipient Rag1−/− mice
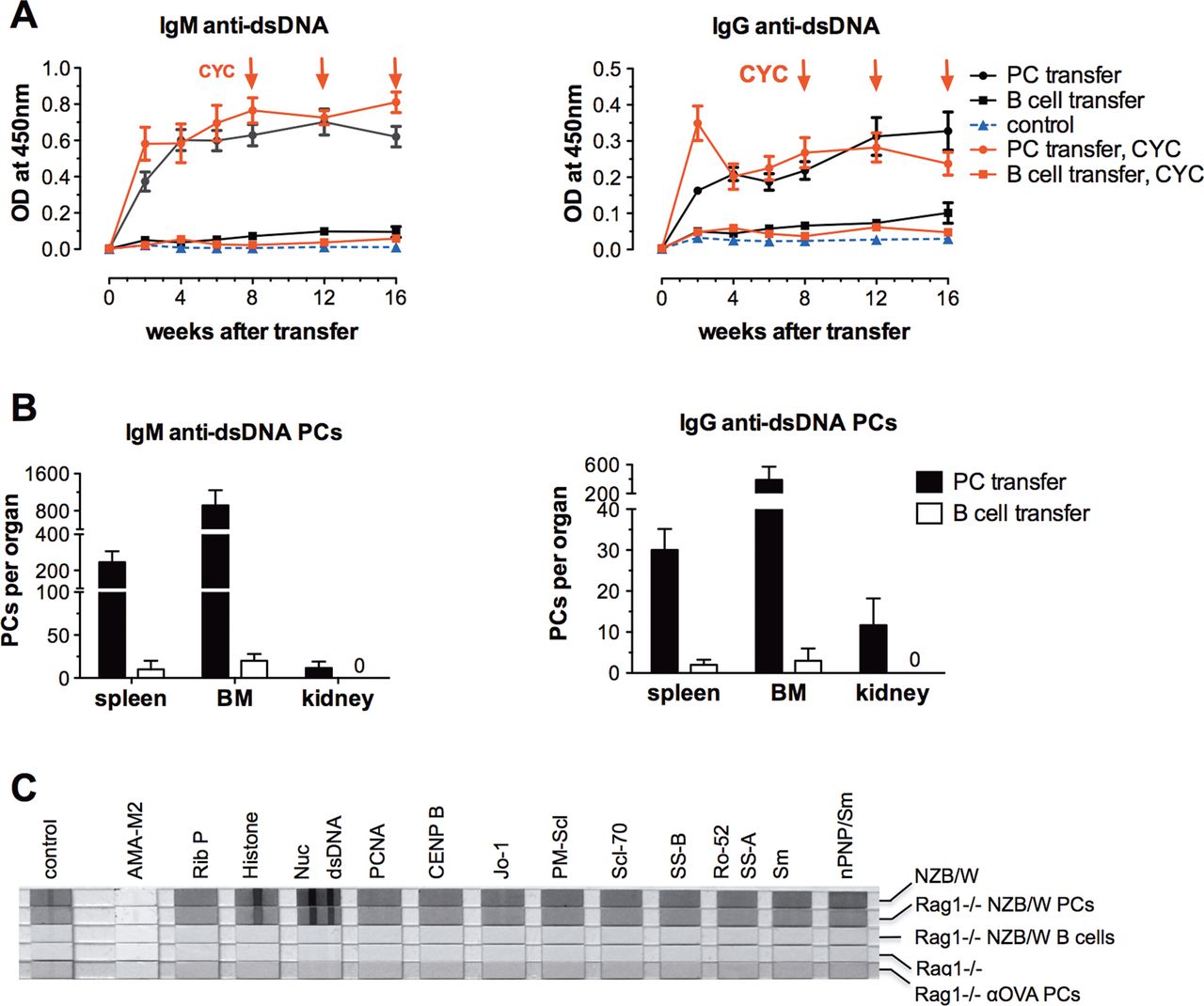
Only 1 week after the transfer of ASCs from NZB/W mice, IgG, IgM and anti-dsDNA levels were detectable in the serum of recipient mice. There was no significant change in antibody levels during the entire observation period of 16 weeks (figure 3A, 4A). The recipients also developed anti-erythrocyte antibodies, as NZB/W mice do (not shown). As expected, control Rag1−/− mice without transfer of ASCs did not have significant levels of total IgG, IgM or anti-dsDNA. As a control experiment, we also transferred anti-OVA ASCs from the spleens of Balb/c mice. This resulted in considerable anti-OVA antibody levels in the recipient mice, while anti-dsDNA antibodies could not be detected (figure 3B). The serum autoantibody profile measured by line immunoassay did not differ between the NZB/W mouse donors of splenic ASCs and the recipient mice. The sera from the NZB/W donors and Rag1−/− recipients exhibited the same ANA profile including anti-dsDNA, anti-histone and anti-nucleosome antibodies, but that from recipient mice transferred with anti-OVA ASCs did not (figure 4C).

Serum antibody and autoantibody levels measured by ELISA in recipient Rag1−/− mice at baseline and after adoptive transfer of splenic antibody-secreting cells (ASCs). (A) Total IgM and IgG as well as IgM anti-dsDNA and IgG anti-dsDNA antibody levels in Rag1−/− mice with and without adoptive transfer of splenic ASCs from NZB/W mice. Data represent the mean of 5 mice per group±SEM. (B) Time course of IgG anti-OVA and anti-dsDNA antibodies in Rag1−/− mice after adoptive transfer of splenic ASCs from Balb/c mice immunised with OVA. Data represent the mean of 10 mice per group±SEM. OVA, ovalbumin.

Autoantibody production levels in Rag1−/− mice after adoptive transfer of splenic antibody-secreting cells (ASCs) from NZB/W mice versus adoptive transfer of B cells in numbers relative to B cell counts (contamination) after isolation of splenic ASCs. (A) Course of IgM and IgG anti-dsDNA antibody levels after transfer of ASCs and B220+ B cells. Subgroups of recipient Rag1−/− mice were treated with cyclophosphamide (CYC) at weeks 8, 12 and 16 (red lines) to verify the role of proliferating cells in autoantibody production. Data represent the mean of 5 mice per group±SEM. (B) Absolute numbers of IgM and IgG anti-dsDNA antibody-secreting plasma cells (PCs) in the spleen, bone marrow (BM) and kidney of recipient Rag1−/− mice 21 weeks after adoptive transfer of ASCs and B220+ B cells. Data represent the mean of 5 mice per group±SEM. (C) Serum autoantibody profile of recipient Rag1−/− mice 21 weeks after transfer of ASCs or B220+ B cells compared to a serum pool from 6- to 7-month-old NZB/W mice and control Rag1−/− mice analysed using the line immunoassay. OVA, ovalbumin.
Low-level B cell contamination of adoptively transferred cells did not contribute to autoantibody production
It was previously shown that the transfer of high numbers (5–15×106) of pre-B cells derived from NZB/W mice to SCID mice leads to autoantibody production, proteinuria and nephritis.26 In our study, the proportion of B cell contamination in the population of transferred ASCs was less than 2%. To ensure that this low-level contamination did not contribute to the production of autoantibodies, the corresponding number of B cells was purified from the spleens of NZB/W mice and transferred into Rag1−/− mice. This did not result in additional serum anti-dsDNA antibody production in the recipient mice, and the IgM and IgG anti-dsDNA antibody levels were comparable to those in the control mice (figure 4A). The negative serum anti-dsDNA antibody results after B cell transfer correspond to the ELISPOT data in week 21, showing that negligible numbers of IgM and IgG anti-dsDNA-secreting PCs were present in the spleen and BM of recipient Rag1−/− mice (figure 4B). The line immunoassay also did not show any serum autoantibody positivity in Rag1−/− mice transferred with B cells (figure 4C).
Autoantibodies exclusively secreted by long-lived PCs result in immune complex nephritis in Rag1−/− mice
To investigate whether long-lived PCs transferred from lupus NZB/W F1 mice can cause pathological changes in the kidneys of Rag1−/− mice, proteinuria was monitored. By 21 weeks after transfer, urinalysis showed proteinuria in eight out of 10 recipient mice but in none of the control Rag1−/− mice, including the vehicle group (n=10), the anti-OVA ASC transfer group (n=10), and the B cell transfer group (n=5) (figure 5A). Renal immunofluorescence analysis in Rag1−/− mice with transferred ASCs from NZB/W mice revealed deposition of C1q, C3, IgM, IgG and kappa, which was identical to the depositions seen in 6- to 7-month-old donor NZB/W mice. Immune depositions were not detectable in control Rag1−/− mice (figure 5C). In contrast to nephritic NZB/W mice, infiltrates of inflammatory cells and PCs were not observed in the renal histology of recipient mice transferred with NZB/W ASCs after 21 weeks. The serum levels of 12 pro-inflammatory cytokines did not differ from those in control Rag1−/− mice (data not shown).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Autoantibodies exclusively secreted by long-lived plasma cells (PCs) induce autoimmune pathology. (A) Analysis of proteinuria in Rag1−/− mice 21 weeks after adoptive transfer of splenic antibody-secreting cells (ASCs) (n=10) or B220+ B cells from NZB/W mice (n=5) and after adoptive transfer of splenic ASCs from Balb/c mice after secondary immunisation with ovalbumin (OVA) (n=10) compared to control Rag1−/− mice (n=10). Statistical differences compared to the recipients with transferred ASCs from NZB/W nice were analysed using the Mann–Whitney test. (B) Survival curve of Rag1−/− mice adoptively transferred with ASCs from NZB/W mice (n=5) compared to that of Rag1−/− mice adoptively transferred with appropriate numbers of B220+ B cells corresponding to B cell contamination levels in the transferred ASC group (n=5) and Rag1−/− control group (n=10) (p=0.0002; Kaplan–Meier log-rank test). (C) Immunohistology of renal sections from Rag1−/− mice adoptively transferred with ASCs from NZB/W mice compared to Rag1−/− control mice, Rag1−/− mice adoptively transferred with low numbers of B220+ B cells corresponding to B cell contamination levels in the transferred ASC group, and Rag1−/− mice adoptively transferred with ASCs from Balb/c mice immunised with OVA 21 weeks after transfer. A renal section from a 7-month-old NZB/W mouse was used as the positive control.
Starting from week 20 after transfer, the Rag1−/− mice transferred with ASCs from NZB/W mice showed reduced survival (figure 5B). At the end of follow-up (week 28), only 20% of these mice had survived, while all of the control Rag1−/− mice were alive.
Discussion
In this study, we have demonstrated for the first time that autoantibodies secreted by long-lived PCs are pathogenic and major drivers of inflammation. They cause immune complex nephritis and reduce survival. We adoptively transferred 2–3×106 ASCs from the spleens of NZB/W mice into Rag1−/− mice. Based on the concept that PBs can migrate but PCs can not,6 ,23 it was expected that PBs, which make up about 70% of all splenic ASCs in NZB/W mice, would migrate to survival niches in the BM and spleen to become long-lived and continuously secrete antibodies, including anti-dsDNA. The attraction of PBs expressing CXCR4 through CXCL12 seems to be the key homing mechanism since the pre-incubation of PBs with the CXCR12 blocker AMD310027 resulted in a reduction in PB homing of more than 70% in recipient Rag1−/− mice (manuscript in preparation). By analysing the intensity of CFSE labelling at different time points, we showed that about 70% of the surviving ASCs underwent two to three cell divisions within the first 14 days after transfer, which agrees with in vitro data.28 Later cell divisions were not observed, indicating that then only mature long-lived PCs secreted antibodies. The proliferation of two-thirds of ASCs increased the number of ASCs in the BM so that it was twice as high on day 30 as on day 3 after transfer. It also indicates that the ASCs surviving the transfer procedure had not occupied all vacant survival niches. Therefore, the remaining free niches were available for PCs arising from proliferating PBs.
The results of BrdU feeding for 2 weeks before sacrifice at week 21 confirmed that non-proliferating long-lived PCs were the only cells responsible for antibody production. Additionally, the contribution of low-level B cell contamination of the transferred cells to the continuous generation of new PCs and to significant (auto)antibody production was excluded by transferring the corresponding number of B cells from the spleens of NZB/W mice into Rag1−/− mice. Finally, cyclophosphamide treatment of recipient Rag1−/− mice that affects proliferating PBs and short-lived PCs5 ,17 did not result in a significant reduction in (auto)antibody levels. Consequently, the increase in proteinuria and the deposition of IgG, IgM, C1q and C3 in the kidneys of the recipient Rag1−/− mice was caused by (auto)antibodies secreted by long-lived PCs. We assume that the deposition was mainly mediated by anti-dsDNA and anti-nucleosome antibodies known to be involved in the pathogenesis of lupus nephritis,29–32 but the contribution of other nephritic autoantibodies not investigated in the serum cannot be excluded. The observed renal immune complex deposition was definitely related to autoantibodies secreted by PCs from NZB/W mice, since the transfer of splenic anti-OVA ASCs into Rag1−/− mice did not lead to similar immunohistological changes in the kidneys.
Notably, there was preferential homing of autoreactive IgG anti-dsDNA-secreting PCs into the BM, which is the main site of long-lived PCs. The detailed mechanisms responsible for this observation, including the migratory and niche-specific mechanisms of migration, remain unclear and should be investigated in future studies. However, it is known that mainly IgG-secreting PCs are long-lived.6
The findings clearly show that the adoptive transfer of ASCs from autoimmune mice into Rag1-deficient mice can result in the development of an exclusively long-lived PC compartment in the BM and spleen that drives autoantibody-mediated inflammation. This mechanism is independent of inflammatory processes accelerated by short-lived autoreactive PBs and PCs,11 which are responsive to antiproliferative drugs12 and treatments targeting B cells.15 As the autoreactive memory PC compartment is in contrast refractory to these treatments,5 ,15 ,17 this mechanism should be considered for future therapeutic strategies in antibody-mediated diseases.
References
Supplementary materials
Supplementary Data
This web only file has been produced by the BMJ Publishing Group from an electronic file supplied by the author(s) and has not been edited for content.
Files in this Data Supplement:
- Data supplement 1 - Online figure
Footnotes
-
Handling editor Tore K Kvien
-
BFH and FH contributed equally.
In memory of our co-author IM, who died too early.
-
Contributors QC, IM and LK carried out most of the experiments. FH, AR, BFH and QC developed the concept, designed the experiments, analysed the data and wrote the manuscript.
-
Funding The work was supported by the Deutsche Forschungsgemeinschaft (SFB650, TP17). LK received a grant from the Berlin-Brandenburg School for Regenerative Therapies.
-
Competing interests None.
-
Provenance and peer review Not commissioned; externally peer reviewed.