Article Text

Statistics from Altmetric.com
Invariant natural killer T (iNKT) cells belong to a prototypical regulatory T cell lineage that functions at the interface of innate immunity and adaptive immunity. Unlike peptide-restricted T cells, iNKT cells recognise and respond to various glycolipid antigens presented in the context of the major histocompatibility complex class I-like molecule CD1d.1 iNKT cells possess a number of unique effector functions, including the ability to rapidly secrete large quantities of Th1, Th2 and Th17 cytokines, and, as such, have had a number of regulatory functions assigned to them. Consequently, iNKT cells are able to regulate a large number of bystander cells that form part of the innate and adaptive immune systems, and iNKT cell biology has subsequently been linked to a broad range of diseases, including cancer, certain infections (eg, Lyme disease) and various autoimmune disorders (eg, rheumatoid arthritis, systemic lupus erythematosus). However, many studies have shown that several mechanisms of iNKT cell activation exist. These differential mechanisms control the mode of activation, the nature of the stimulating glycolipid, genetic background or even the timing of iNKT cell activation. As such, the outcome of experimental models of autoimmunity has been shown to range from protection to disease aggravation, the findings of which have led to a matter of debate as to the precise role of these cells in autoimmune diseases (for reviews, see Coppieters et al2 and Drennan et al3). In this context, the identification of factors that influence the outcome of iNKT cell effector function has been expanded to include structural modifications of specific glycolipid antigens that promote T helper cell cytokine bias, types of antigen-presenting cells (APCs), subsets of the iNKT cells themselves and the role of costimulatory molecules (for a review, see Van Kaer et al4).
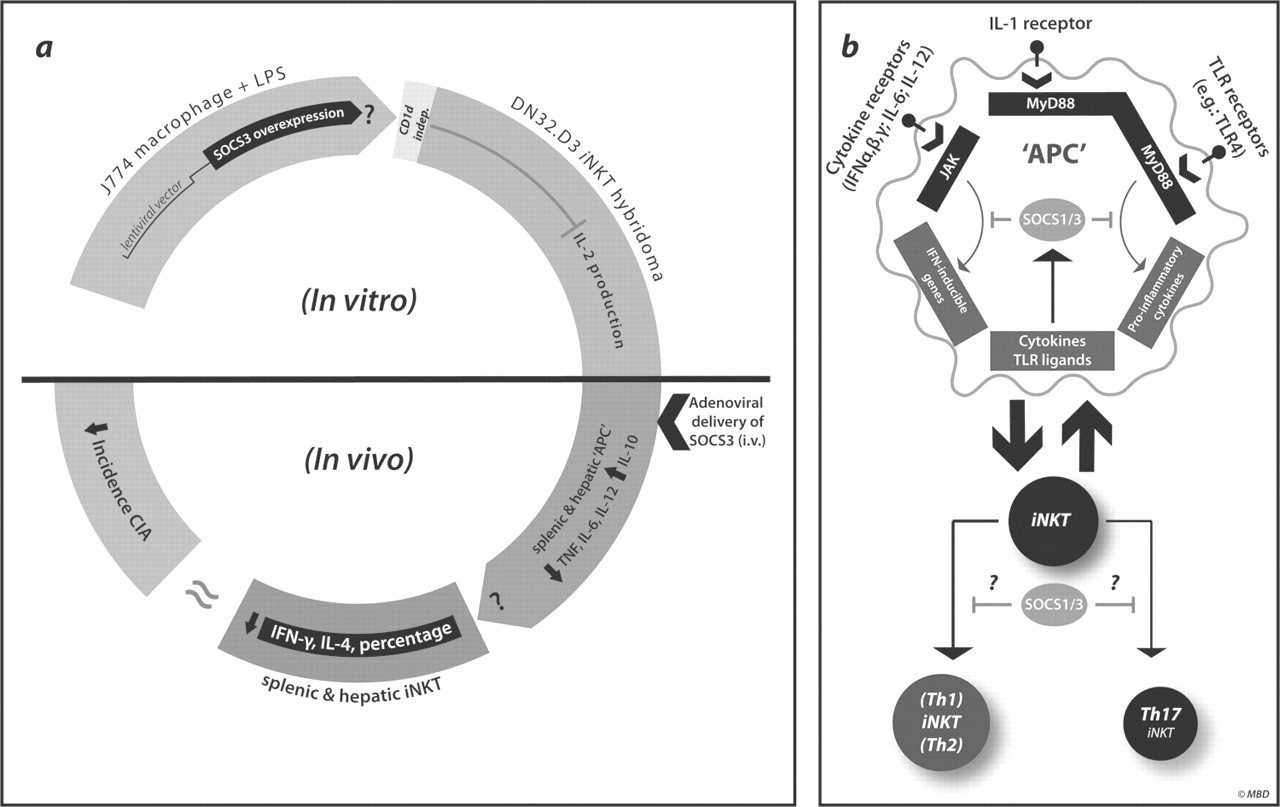
Although several studies have highlighted the modular nature of iNKT cell activation, surprisingly little is known about the potential negative regulators of iNKT cell function. Hypothetically speaking, negative regulators of iNKT cell activation could function either within a cell-intrinsic fashion, indirectly by influencing APC activation or via a combination of both. Veenbergen et al5 describe a model suggesting that a member of the suppressors of cytokine signalling (SOCS) family, namely SOCS3, may be a negative regulator of iNKT cell function during collagen-induced arthritis in mice (figure 1A). By the use of an adenoviral delivery system, SOCS3 was overexpressed in APCs, and the impact on iNKT cell responses was determined. Here, SOCS3-transduced J774 macrophages dampened iNKT-cell-dependent interleukin (IL)-2 secretion following stimulation with the prototypical iNKT cell ligand α-galactosylceramide. A similar effect was seen when SOCS3-overexpressing macrophages were stimulated with lipopolysaccharide (LPS), a toll-like receptor (TLR) 4 ligand. In addition, in vivo experiments using adenoviral transfer of SOCS3 clearly prevented the development of collagen-induced arthritis. This is in line with earlier studies. Of interest, these adenoviruses target the APC fraction rather than the T cell fraction in vivo. Interestingly, the expression of the early activation marker CD69 in liver or spleen iNKT cells was reduced compared to the expression in mice exposed to a control virus. Furthermore, ex vivo SOCS3-transfected APCs retained the ability to modulate iNKT cell responses to α-galactosylceramide, LPS or a combination of both. These results confirmed their primary in vitro results. Additional results suggested that the effect of SOCS3 was independent of glycolipid presentation in the context of CD1d, yet appeared to be associated with altered cytokine levels such as IL-10. Collectively, these results suggest that SOCS3 may be an important negative regulator of iNKT cell function.

{kind=link}
(A) SOCS3-dependent model proposed by Veenbergen et al. In vitro results suggest that the lentiviral transfection of a J774 macrophage cell line with SOCS3 inhibited IL-2 production by the iNKT cell hybridoma DN32.D3 in response to either LPS or α-galactosylceramide. Here, IL-2 production appeared to be CD1d-independent. Furthermore, the authors suggest that adenoviral delivery of SOCS3 resulted in uptake of SOCS3 by splenic and hepatic ‘APCs’, reduced cytokine production by splenic and hepatic iNKT cells, and moderate protection during a murine model of collagen-induced arthritis. (B) Potential role for SOCS proteins during iNKT cell activation. Cytokines and TLR ligands signal through receptor complexes that activate the JAK/STAT and MyD88 signalling pathways. Activation of downstream transcription factors upregulates SOCS1 and SOCS3 gene expression, which, when translated, serves to produce proteins that suppress TLR, IL-1R and cytokine signalling pathways in a negative feedback loop. It is currently unknown how SOCS1/SOCS3 expression within APCs regulates iNKT cell activation and whether SOCS1/SOCS3 have cell-intrinsic functional roles in iNKT cell cytokine production, as a role for these proteins has been described for T helper cell lineage specification within the conventional T cell pool (for a review, see Palmer and Restifo8). SOCS, suppressors of cytokine signalling; IL, interleukin; iNKT, invariant natural killer T; LPS, lipopolysaccharide; APC, antigen-presenting cell; TLR, toll-like receptor.
The results from this study extend earlier work on SOCS family members regulating iNKT cell function. Here, SOCS1 deficiency, also termed STAT (signal transducer and activator of transcription)-induced STAT inhibitor-1, was found to lead to fulminant hepatitis.6 Deletion of iNKT cells in SOCS1-deficient animals substantially reduced the hepatitis, suggesting an intrinsic negative feedback loop exerted by SOCS1. More recently, with the use of a transgenic approach, forced expression of SOCS3 in T and iNKT cells suppressed concanavalin-A-induced hepatitis by inhibiting iNKT-cell-dependent cytokine production.7 Conversely, conditional knockout mice specific for T and iNKT cells were hypersensitive to autoimmune hepatitis. Overall, these results underscore the relevance of both SOCS1 and SOCS3 to modulating iNKT cell function, either directly or indirectly, by influencing APCs. Of peculiar interest is the intrinsic role of these negative regulators under homeostatic conditions. This raises some important questions that remain to be resolved: (a) How is SOCS3 regulated in the iNKT cells themselves, and what are the potential upstream signalling pathways? (b) Are there differences in SOCS3-mediated suppression in distinct iNKT cell subsets or in different types of APC (figure 1B)? Furthermore, it is not clear whether these inhibitory pathways are altered under conditions of chronic inflammation such as in inflamed joints. It should be highlighted that SOCS3 affects several cell types; thus, clearly, the contribution of SOCS3 to the modulation of iNKT cells as a means to downmodulate experimental arthritis remains to be proven. Hence, given the well-established role of SOCS3 in dampening mainstream T-cell-dependent responses, it seems likely that this may be an equally important mode of action. Thus, clearly more research is needed to determine whether the discovery of SOCS proteins as negative regulators of regulatory cell types is only the tip of the iceberg. It could be envisioned that a whole range of negative regulatory feedback loops exist, with even some selectivity according to lymphocyte subsets. The emerging role of SOCS proteins in modulating iNKT cell function, together with the additional mechanisms uncovered to fine-tune the function of these cells, highlights their critical role in immune homeostasis in health and disease.
Footnotes
-
Competing interests None.
-
Provenance and peer review Commissioned; externally peer reviewed.