Article Text

Abstract
Objectives Synovial fibroblasts and osteoblasts generate active glucocorticoids by means of the 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) enzyme. This activity increases in response to proinflammatory cytokines or glucocorticoids. During inflammatory arthritis synovium and bone are exposed to both these factors. This study hypothesised that glucocorticoids magnify the effects of inflammatory cytokines on local glucocorticoid production in both synovium and bone.
Methods The effects of inflammatory cytokines (IL-1β/tumour necrosis factor alpha; TNFα) and glucocorticoids, alone or combined, were assessed on the expression and activity of 11β-HSD1 in primary synovial fibroblasts, primary human osteoblasts and MG-63 osteosarcoma cells. A range of other target genes and cell types were used to examine the specificity of effects. Functional consequences were assessed using IL-6 ELISA.
Results In synovial fibroblasts and osteoblasts, treatment with cytokines or glucocorticoids in isolation induced 11β-HSD1 expression and activity. However, in combination, 11β-HSD1 expression, activity and functional consequences were induced synergistically to a level not seen with isolated treatments. This effect was seen in normal skin fibroblasts but not foreskin fibroblasts or adipocytes and was only seen for the 11β-HSD1 gene. Synergistic induction had functional consequences on IL-6 production.
Conclusions Combined treatment with inflammatory cytokines and glucocorticoids synergistically induces 11β-HSD1 expression and activity in synovial fibroblasts and osteoblasts, providing a mechanism by which synovium and bone can interact to enhance anti-inflammatory responses by increasing localised glucocorticoid levels. However, the synergistic induction of 11β-HSD1 might also cause detrimental glucocorticoid accumulation in bone or surrounding tissues.
This paper is freely available online under the BMJ Journals unlocked scheme, see http://ard.bmj.com/info/unlocked.dtl
Statistics from Altmetric.com
Inflammatory joint disease is associated with focal bone loss, periarticular osteopenia and generalised osteoporosis. Bone loss is associated with uncoupling of bone resorption from formation but the basis for this is poorly understood.1 2 Systemic bone loss in other inflammatory diseases is exaggerated by treatment with therapeutic glucocorticoids.3 How inflammation sensitises bone to glucocorticoids is unknown. We have previously shown that synovial fibroblasts and osteoblasts generate active glucocorticoids through the expression of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1).4,–,7 This enzyme converts inactive glucocorticoids (cortisone and prednisone) to their active counterparts (cortisol and prednisolone). This activity increases in vitro in response to proinflammatory cytokines or glucocorticoids.4 5 8 Glucocorticoid generating capacity in synovial tissue correlates with the erythrocyte sedimentation rate in patients with inflammatory arthritis.6 Systemic measures of 11β-HSD1 activity also predict the effect of therapeutic glucocorticoids on bone markers in healthy men.9 However, in clinical situations both proinflammatory cytokines and glucocorticoids are likely to impact on 11β-HSD1 expression but the effect of these factors in combination is unclear. Typically, glucocorticoids inhibit proinflammatory cytokine signalling10 but the presence of high 11β-HSD1 activity in inflamed joints despite high glucocorticoid levels suggested that glucocorticoids fail to suppress the effect of proinflammatory cytokines on 11β-HSD1. We examined this hypothesis in primary synovial fibroblasts and osteoblasts. We found that 11β-HSD1 expression and activity increased dramatically in response to the combination of inflammatory cytokines and glucocorticoids. This induction was specific for 11β-HSD1, did not occur in foreskin fibroblasts or adipocytes, and had functionally important effects.
Methods
Cell culture
Reagents were obtained from Sigma (Poole, UK) unless stated. Primary synovial fibroblasts were generated from synovial tissue obtained from patients with rheumatoid arthritis (by 1987 American Rheumatism Association classification criteria) during knee arthroplasty as described.4 Isolated fibroblasts were grown in RPMI-1640 medium containing 1% (v/v) non-essential amino acids (NEAA), 1% penicillin/streptomycin, 1% sodium pyruvate, 2mM glutamine and 20% fetal bovine serum (FBS; Labtech, Sussex, UK). Hs-68 cells, a primary culture of human foreskin fibroblasts, were cultured in Dulbecco's modified Eagle's medium (DMEM) with 20% FBS and 2mM glutamine.
Primary human osteoblasts were cultured from bone chips obtained at orthopaedic surgery as described.8 Trabecular bone was treated with 2% type 1 collagenase for 2 h, washed then cultured in DMEM/F12 containing 10% FBS, 1% NEAA and 2 mmol/litre glutamine. MG-63 cells were cultured in minimal essential medium with 10% FBS, 1% NEAA and 2 mmol/litre glutamine. SV-HFO cells were cultured in α minimal essential medium (Gibco, Paisley, UK) with 20 mmol/litre HEPES, pH 7.5, streptomycin/penicillin, 1.8 mmol/l calcium chloride, 2% FBS and 10 mmol/litre β-glycerophosphate.
Chub-S7 cells, a human pre-adipocyte cell line, were cultured in DMEM/F12 with 10% FBS to maintain a preadipocyte phenotype. Treatment with DMEM/F12 with 10% FBS, 17µM D-panthotenic acid, 33µM biotin, 1nM triiodothyronine, 166nM insulin, 1µM cortisol and 1µM GW1929 was used to generate mature adipocytes. Patients gave informed consent and the study had Local Research Ethics Committee approval.
Cells were cultured with proinflammatory cytokines (IL-1β/tumour necrosis factor alpha (TNFα); 0.1–10 ng/ml) and glucocorticoids (cortisol/dexamethasone; 1–100 nM) alone or in combination.
RNA extraction and real-time PCR
RNA was extracted using TRI reagent (Sigma). Aliquots (1 µg) were reverse transcribed using random hexamers (Promega, Madison, Wisconsin, USA).5 The expression of messenger RNA for 11β-HSD1, 11β-HSD2, GRα, GRβ, osteoprotegerin and cyclooxygenase-2 was assessed using real-time PCR using an ABI7500 system (Applied Biosystems, Warrington, UK) with 18S as internal reference. Reactions contained TaqMan PCR mastermix, 500–900 nmol primers, 100–200 nmol TaqMan probe and 25–50 ng complementary DNA. Data were obtained as Ct values and used to determine ΔCt values (Ct target – Ct 18S). Fold-change in expression was determined by subtracting ΔCt values for treated cells from their control. The resulting ΔΔCt values were used to calculate fold-change using the equation 2ΔΔCt. Probe and primer sequences were as follows: 11β-HSD1, forward primer AGGAAAGCTCATGGGAGGACTAG, reverse ATGGTGAATATCATCATGAAAAAGATTC, probe CATGCTCATTCTCAACCACATCACCAACA; GRα, forward AACTGGCAGCGGTTTTATCAACT, reverse AATACTCATGGTCTTATCCAAAAATGTTT, probeATTCTATGCATGAAGTGGTGGAAAATCTCCTTAACTATTG; GRβ forward AACTGGCAGCGGTTTTATCAACT, reverse TGTGTGAGATGTGCTTTCTGGTT, probe AACTCTTGGATTCTATGCATGAAAATGTTATGTGGTTA. TaqMan assays for osteoprotegerin (Hs00171068_m1) and cyclooxygenase-2 (Hs00153133_m1) were from Applied Biosystems.
Western blot analysis
The expression of 11β-HSD1 protein was determined by western blotting using a specific 11β-HSD1 antibody (Binding Site, UK).11 Expression was analysed relative to β-actin. Densitometry was performed on three separate experiments using primary synovial fibroblasts from three different individuals.
11β-HSD enzyme activity assays
Confluent cells were cultured in medium containing 100 nM cortisone (to measure oxo-reductase/activation activity) or cortisol (dehydrogenase/inactivation activity) along with tritiated tracer. Steroids were extracted using dichloromethane and separated by thin-layer chromatography using ethanol:chloroform (8:92) as the mobile phase. Thin-layer chromatography plates were analysed by Bioscan imager (Bioscan, Washington, DC, USA) and the fractional conversion of steroids was calculated. The protein concentration was assessed by a 96-well assay kit (Bio-Rad, Hercules, California, USA). Results were expressed as pmol product/h per milligram of protein and experiments were performed in triplicate.
Functional consequences of induction of 11β-HSD1 activity on IL-6 production
Primary fibroblasts were incubated with no treatment, 10 ng/ml IL-1β, 100 nmol/litre dexamethasone or both for 24 h. Medium was then changed to fresh medium without cytokines or glucocorticoids and left for 24 h. Cells were then treated for 24 h with or without 100 nmol/litre cortisone and media collected. Soluble IL-6 was measured by sandwich ELISA (BD Biosciences, Torreyana, San Diego, California, USA). Data were expressed as the production of IL-6 in pg/well.
Statistics
Data were reported as mean±standard error (SE) of replicate mean values. One-way and two-way analysis of variance analyses with Tukey post-hoc test were performed using SPSS Data Editor and SigmaStat software.
Results
Inflammatory cytokines and glucocorticoids synergistically induce 11β-HSD1 expression and activity in synovial fibroblasts
As previously reported, when used in isolation, pro-inflammatory cytokines (IL-1β/TNFα) and glucocorticoids increased 11β-HSD1 mRNA expression and oxidoreductase enzyme activity in primary human synovial fibroblasts (figure 1A,B). However, the combination of IL-1β and glucocorticoids substantially increased 11β-HSD1 mRNA levels and enzyme activity relative to either individual treatment. This pattern of activity was consistently seen in cells from three separate donors and data reflect the combined results from these three patients. In keeping with our previous report, no dehydrogenase activity was detectable in these cells.4 Western blotting using extracts from primary synovial fibroblasts showed that this change was caused by increased 11β-HSD1 protein expression (figure 1C).

Synergistic induction of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) mRNA, activity and protein expression in synovial fibroblasts by inflammatory cytokines and glucocorticoids. (A) Effect of 10 ng/ml IL-1β/tumour necrosis factor alpha (TNFα), 100nM dexamethasone (DEX) or combined IL-1β/TNFα and dexamethasone on the expression of 11β-HSD1 mRNA expression as determined by real-time reverse transcriptase PCR. *p<0.05, **p<0.01 compared with control (Con); #p<0.05 compared with single treatments. Data are based on cultures from three individuals. (B) Effect of 10 ng/ml IL-1β/TNFα, 100nM dexamethasone or combined IL-1β/TNFα and dexamethasone on 11β-HSD1 catalysed synthesis of 3H-cortisol from 3H-cortisone (oxo-reductase activity) as determined by scanning thin-layer chromatography. Data are shown as pmoles/h per milligram of protein and are based on cultures from three individuals. (C) Effect of IL-1β, dexamethasone or combined IL-1β/dexamethasone on 11β-HSD1 protein expression in primary cultures of synovial fibroblasts as determined by western blotting. Expression is relative to β-actin. Densitometry data reflect expression in three separate primary cultures of synovial fibroblasts derived from different patients.
Inflammatory cytokines and glucocorticoids synergistically induce 11β-HSD1 in osteoblasts
The same pattern of induction of 11β-HSD1 mRNA and enzyme activity was also observed in primary human osteoblasts and MG63 cells (figure 2A,B). Similar results were obtained using TNFα as co-treatment (for MG-63 cells increases in mRNA expression relative to untreated cells: 54-fold with 10 ng/ml TNFα; 19-fold with 100 nM dexamethasone; 2952-fold with combined TNFα/dexamethasone), or when using cortisol as co-treatment (increases in mRNA expression: 12-fold with 100 nM cortisol; 1150-fold with combined TNFα/cortisol).
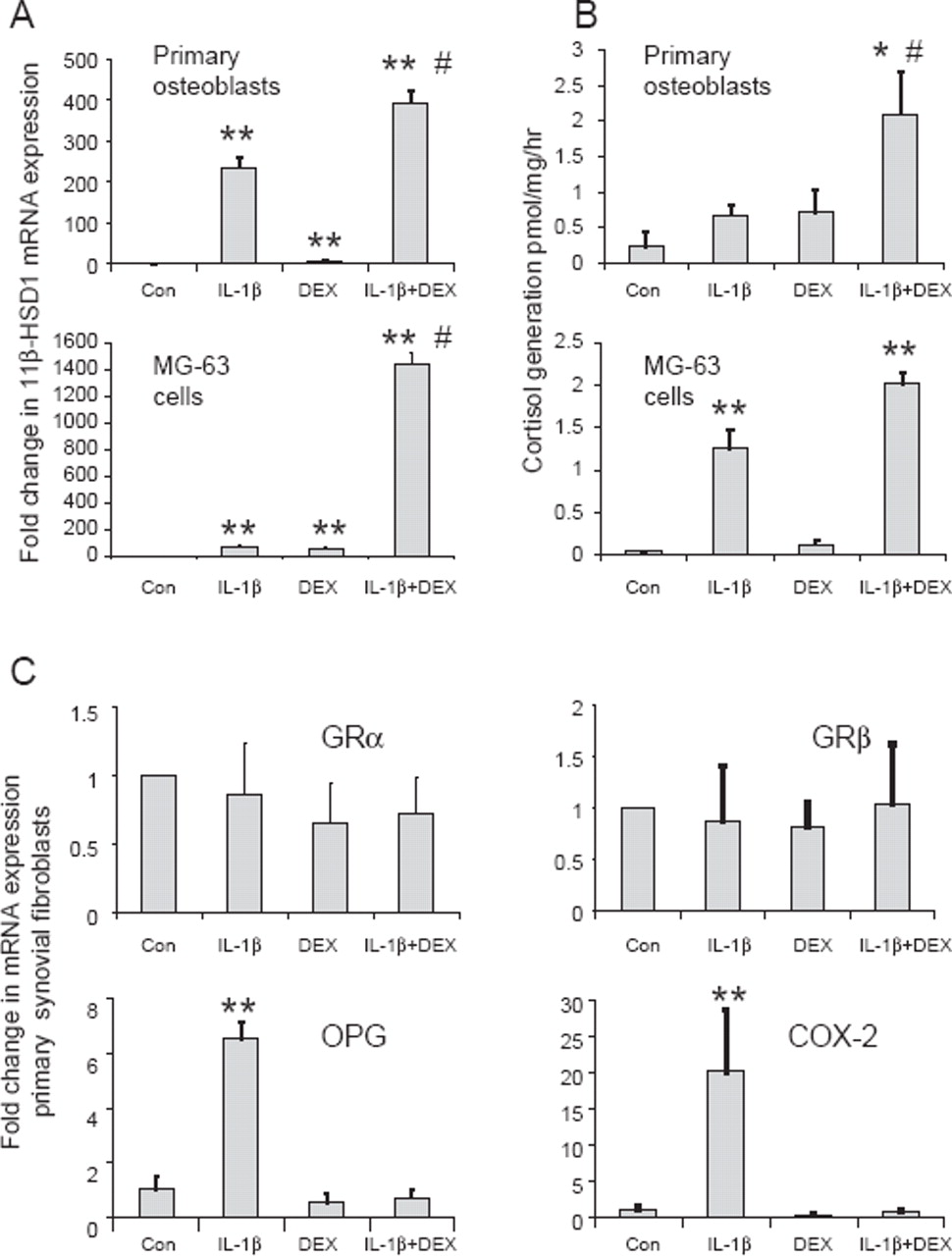
Synergistic induction of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) mRNA and activity in human osteoblastic cell culture models by inflammatory cytokines and glucocorticoids. (A) Effect of 10 ng/ml IL-1β, 100nM dexamethasone (DEX) or combined IL-1β/dexamethasone on the expression of 11β-HSD1 mRNA in primary human osteoblasts and MG-63 cells as determined by real time reverse transcriptase PCR. Data are normalised for 18S rRNA expression and are shown as fold-change relative to an arbitrary value of one for vehicle-only control cells (Con). *p<0.05, **p<0.01 compared with control; #p<0.05 compared with single treatments, N=3. (B) Effect of 10 ng/ml IL-1β, 100nM dexamethasone and combined IL-1β/dexamethasone on 11β-HSD1 catalysed synthesis of 3H-cortisol from 3H-cortisone (oxo-reductase activity) by primary human osteoblasts and MG-63 cells as determined by scanning thin-layer chromatography. Data are shown as pmoles/h per milligram of protein, N=3. (C) Effects of combined IL-1β and dexamethasone on expression of mRNA for GRα, GRβ, osteoprotegerin (OPG) and cyclooxygenase-2 (COX-2) in primary synovial fibroblasts demonstrating that synergism is specific for the induction of 11β-HSD1 expression. Data are based on cultures from three individuals.
Synergistic response to inflammatory cytokines and glucocorticoids is specific for 11β-HSD1
Analyses using primary synovial fibroblasts confirmed that a synergistic effect of co-treatment with IL-1β and dexamethasone was not observed for other glucocorticoid signalling components such as GRα/β (figure 2C). Moreover, analysis of two cytokine gene targets, osteoprotegerin and cyclooxygenase-2, showed that although the expression of both was increased following IL-1β treatment, this effect was abrogated rather than potentiated in combination with dexamethasone (figure 2C).
Synergistic induction of 11β-HSD1 expression occurs in a limited number of cell types
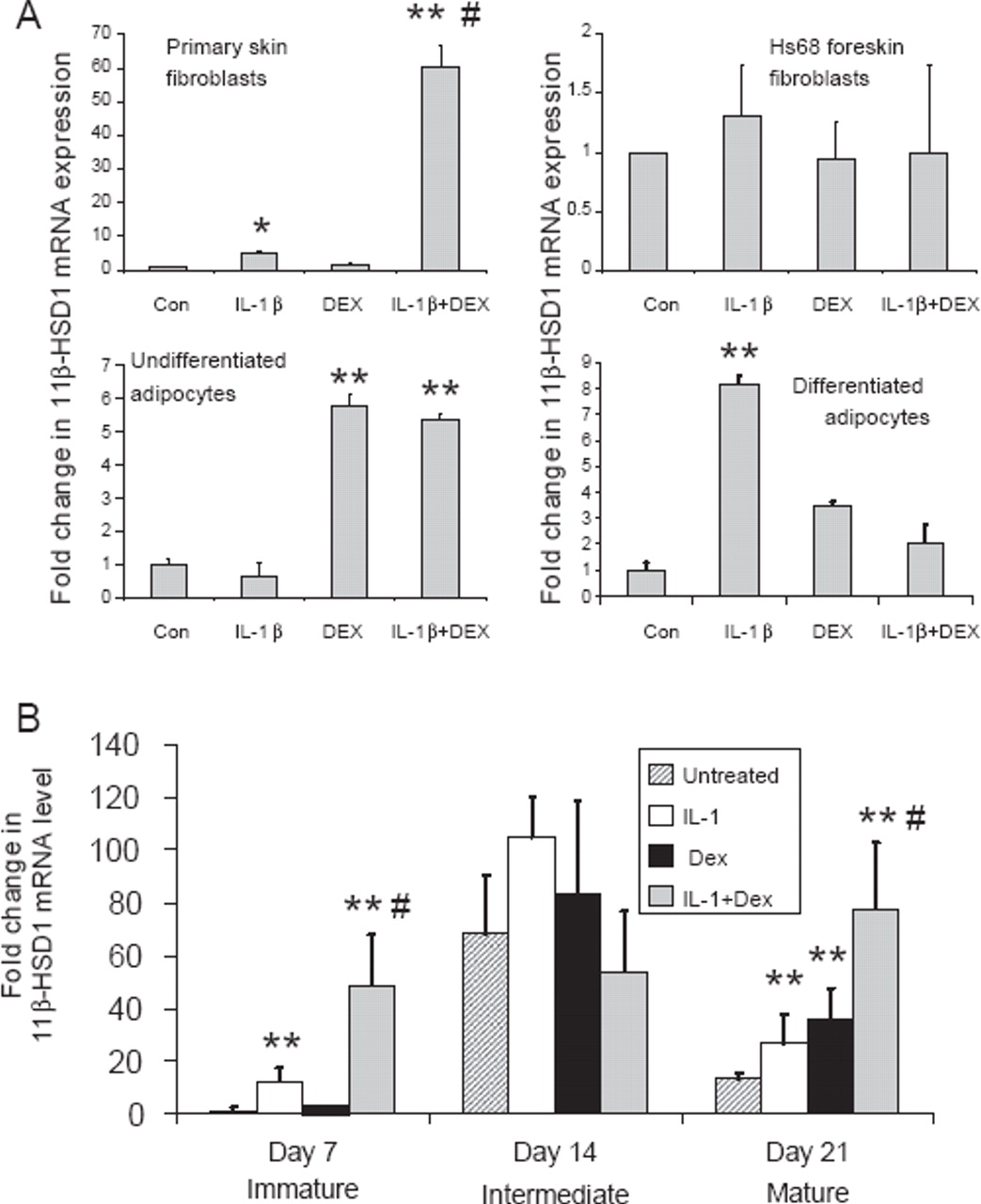
The effects of IL-1β and dexamethasone were examined in other cell types. A synergistic effect on 11β-HSD1 expression was seen in primary skin fibroblasts isolated from the patients from whom synovial fibroblasts were generated (figure 3A). This effect was not, however, observed in Hs68 primary fibroblasts derived from foreskin. Adipocytes and preadipocytes express 11β-HSD1, and expression is increased by pro-inflammatory cytokines or glucocorticoids.12 13 The effect of treatments in isolation or combination was examined in Chub-S7 human differentiating adipocytes (figure 3A). Glucocorticoid treatment alone induced 11β-HSD1 expression in undifferentiated cells with no effect of IL-1β or any additional effect of IL-1β in combination with dexamethasone evident. In differentiated adipocytes, IL-1β and dexamethasone in isolation had a small impact on 11β-HSD1 mRNA expression, but the effect of the combination was less than either treatment alone. This indicates that synergistic interactions are not universal in cells expressing 11β-HSD1 activity or of stromal origin.

Synergistic effect of IL-1β and dexamethasone (DEX) is restricted to fibroblasts and osteoblasts and depends on the stage of osteoblast differentiation. (A) Effects of combined IL-1β and dexamethasone on the expression of mRNA for 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) in primary skin fibroblasts, foreskin fibroblasts, undifferentiated and differentiated adipocytes. *p<0.05 and **p<0.01 compared with control (Con); #p<0.05 compared with single treatments, N=3. (B) Synergistic induction of 11β-HSD1 occurs at different stages of osteoblast differentiation. Human osteoblastic SV-HFO cells were cultured for 7, 14 or 21 days under the following conditions: control cells (vehicle); cells + 10 ng/ml IL-1β; cells + dexamethasone for 24 h; cells + IL-1β + dexamethasone for 24 h. At each time point RNA was isolated for real-time reverse transcriptase PCR analysis. Data are shown as the mean±SE fold-induction in 11β-HSD1 mRNA expression relative to day 7 cells. **p<0.01 compared with untreated control at each time point; #p<0.05 compared with single treatments, N=6.
Synergistic induction of 11β-HSD1 varies with osteoblast differentiation
To investigate whether the synergistic induction of 11β-HSD in osteoblasts occurs at specific stages of osteoblast differentiation, we assessed the effects of IL-1β and dexamethasone on 11β-HSD1 expression in SV-HFO human fetal osteoblasts. In previous studies we showed that cells readily express 11β-HSD1 mRNA and enzyme activity when cultured without glucocorticoids, and that this activity aids the mineralisation of mature cultures. However, when treated with 100 nM dexamethasone, cells lose the expression of 11β-HSD1, suggesting that this pathway becomes redundant in the presence of exogenous dexamethasone.14 15 Data in figure 3B confirmed previously reported observations, that cells show an increase in 11β-HSD1 expression between days 7 and 14 of culture, with a subsequent decline at day 21. In cells with low 11β-HSD1 expression (days 7 and 21 cultures) treatment with either IL-1β (10 ng/ml) or dexamethasone (100 nM) for the last 24 h of culture induced the expression of 11β-HSD1. As with other osteoblast models, this response was enhanced when IL-1β and dexamethasone were added together. Cells at day 14 did not show altered 11β-HSD1 expression with single or combined treatments, but basal expression was already as high as the maximal expression seen at other times with combined treatments. SV-HFO cells exposed to continual dexamethasone treatment showed suppressed 11β-HSD1 expression at all time points but high expression when cells were co-treated with IL-1β for 24 h (data not shown).
Dose response and time course of 11β-HSD1 induction
Dose-dependency studies of MG-63 cells indicated that synergistic interaction between IL-1β and dexamethasone occurred at all tested concentrations of IL-1β (figure 4A). Synergistic induction of 11β-HSD1 was also seen across a range of concentrations of dexamethasone (1–100 nM; data not shown). The effect of combined IL-1β with dexamethasone compared with IL-1β alone was seen at 8 h, and the difference was most dramatic at 24–48 h (figure 4B, expression of dexamethasone alone not shown because the relative expression was very low).

Dose response and time course of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) induction. (A) Dose-response effects of 10 ng/ml IL-1β, 100 nM dexamethasone (DEX) and combined IL-1β and dexamethasone on 11β-HSD1 mRNA expression in MG-63 cells. Combined treatment at 0.1 ng/ml and all treatments at 1 and 10 ng/ml significantly induced expression compared with untreated controls (p<0.05). #p<0.05 compared with effects of single treatments at each cytokine concentration, N=3. (B) Time course of induction of 11β-HSD1 mRNA following treatment of MG-63 cells with IL-1β or IL-1β + dexamethasone. All treatments significantly induced expression compared with time 0 (p<0.01). #p<0.05 compared with effects of single treatments at each time point, N=3.
Functional impact of synergistic induction of 11β-HSD1 activity in primary fibroblasts
The functional impact of the synergistic induction of 11β-HSD1 activity was examined in synovial fibroblasts treated with IL-1β, dexamethasone or a combination of IL-1β and dexamethasone for 24 h followed by 24 h without treatment (washout period). Cells were then cultured for 24 h with or without 100 nM cortisone before media were removed for the analysis of IL-6 levels. Under these conditions the addition of IL-1β or dexamethasone in isolation did not significantly sensitise cells to cortisone (figure 5). However, cells treated with the combination of IL-1β and dexamethasone were markedly responsive to cortisone, as evidenced by a significant reduction in IL-6 production.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Functional impact of 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) synergism in primary synovial fibroblasts. The functional response to the synergistic induction of 11β-HSD1 activity by IL-1β and dexamethasone (DEX) was assessed in primary synovial fibroblasts. Cells were treated for 24 h with either IL-1β, dexamethasone or combined IL-1β and dexamethasone. Medium was then removed and cells were treated for a washout period of 24 h with unsupplemented medium. The functional impact of differences in 11β-HSD1 expression was then examined by treating cells for 24 h with cortisone (inactive substrate for 11β-HSD1). Data are shown as pg IL-6 produced during this 24 h incubation period. Data are based on cultures from three individuals.
Discussion
We found that the exposure of synovial fibroblasts or osteoblasts to both proinflammatory cytokines and glucocorticoids dramatically increased glucocorticoid-generating capacity relative to each factor in isolation. This synergistic induction of glucocorticoid-generating capacity had important functional consequences. This potentiation, rather than suppression, of localised inflammation-induced expression of 11β-HSD1 by glucocorticoids may be an endogenous mechanism to reduce synovitis. However, with persistent inflammation, the resulting tissue-specific elevation of glucocorticoid generation is likely to contribute to periarticular bone loss in inflammatory joint disease, and could explain why inflammatory diseases sensitise bone to the effects of therapeutic glucocorticoids.
There are multiple interactions between pro-inflammatory cytokines and glucocorticoids. In most situations glucocorticoids antagonise the actions and production of pro-inflammatory cytokines through the inhibition of activator protein 1 or nuclear factor kappa B signalling pathways.10 Additive or synergistic interaction is less commonly reported.16,–,18 Synergistic interactions in these situations have been reported to be by effects on p38-mitogen-activated protein kinase, mitogen-activated protein kinase phosphatase or increased mRNA stability. A glucocorticoid–cytokine interaction has previously been reported for 11β-HSD1 in chorionic trophoblast cells.19 On the basis of promoter–reporter assays it was concluded that this effect was transcriptionally mediated by means of the proximal promoter.
Recent reports have demonstrated 11β-HSD1 expression in synovial fibroblasts in vitro and in vivo.4 6 20 Local generation of active glucocorticoids in patients with inflammatory arthritis was demonstrated on the basis of synovial fluid corticosteroid measurements, and this activity correlated with measures of inflammation.6 This increase in local steroid generation might be aimed at resolving prolonged synovial inflammation. It is possible that in transient synovitis, locally produced glucocorticoids reduce inflammation and hasten resolution. However, in rheumatoid arthritis, this increased generation of glucocorticoids does not lead to the complete resolution of inflammation. The reason for this failure is unclear, but the presence of cells within the joint that are resistant to locally produced glucocorticoids is a potential explanation.6 20 21
The mechanisms underlying periarticular bone loss in inflammatory arthritis remain unclear. Increased bone resorption due to increased osteoclast formation has been proposed, and the pharmacological inhibition of bone resorption can prevent bone loss in animal models.22 However, this bone loss is not accompanied by an expected compensatory increase in bone formation. We have hypothesised that this uncoupling of formation from resorption is due to the production of glucocorticoids within periarticular osteoblasts or nearby synovium.5 6 The findings in this study support this hypothesis and also suggest that glucocorticoids generated within the synovium could increase glucocorticoid-generating capacity in periarticular bone in the presence of proinflammatory cytokines. It is also possible that glucocorticoids generated within osteoblasts could have effects on synovial fibroblasts or immune cells within the synovium.
How glucocorticoids potentiate the effects of proinflammatory cytokines on 11β-HSD1 transcription remains unclear. The finding that adipocytes are responsive to glucocorticoid or cytokine induction of 11β-HSD1 expression but not the combination indicates that tissue-specific factors are important. The reason for the variation of induction across osteoblast differentiation also needs to be determined. It is possible that the lack of effect during mid-differentiation is explained by the high expression of 11β-HSD1 at this stage. We have previously shown that induction across differentiation is mediated by the proximal promoter.14 It will be important to characterise the tissue specificity of these effects given that many cell types express 11β-HSD1.11 23,–,25 Our results show clear heterogeneity of this effect across tissues.
The identification of a powerful interaction between pro-inflammatory cytokines and glucocorticoids for the induction of glucocorticoid-generating capacity has implications for bone diseases. It may explain why patients with inflammatory disorders develop more bone loss with glucocorticoids than those without.26 The biological significance of the synergistic induction of 11β-HSD1 is also unclear, but the dynamics of induction suggest that local glucocorticoid production will occur hours to days after exposure to inflammation. This may therefore be a mechanism to downregulate excessive inflammation (akin to the rise in cortisol levels due to hypothalamo–pituitary–adrenal axis activation with stress).27 The observation that only low glucocorticoid concentrations are required to induce 11β-HSD1 synergistically may explain why even low-dose glucocorticoids are linked to bone loss in inflammatory diseases.28 These observations also need to be considered when evaluating the clinical effects of 11β-HSD1 enzyme inhibitors.29 On the basis of this study some effects of these drugs may only become apparent in the presence of both inflammation and the levels of glucocorticoids seen during stress.
Acknowledgments
The authors would like to thank Laura Gathercole for help with western blot techniques.
References
Footnotes
KK and RH contributed equally.
-
Funding This study was funded by the Medical Research Council, UK and the Arthritis Research Campaign Project grants 17730 and 18081.
-
Competing interests None.
-
Patient consent Obtained.
-
Ethics approval This study was conducted with the approval of the South Birmingham Research Ethics Committee.
-
Provenance and peer review Not commissioned; externally peer reviewed.